Molecular-Clinical Correlation in Pediatric Medulloblastoma: A Cohort Series Study of 52 Cases in Taiwan

 , , ,
, , ,  , , , , , , , add
Show full author list
, , , , , , , add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Patient Cohort
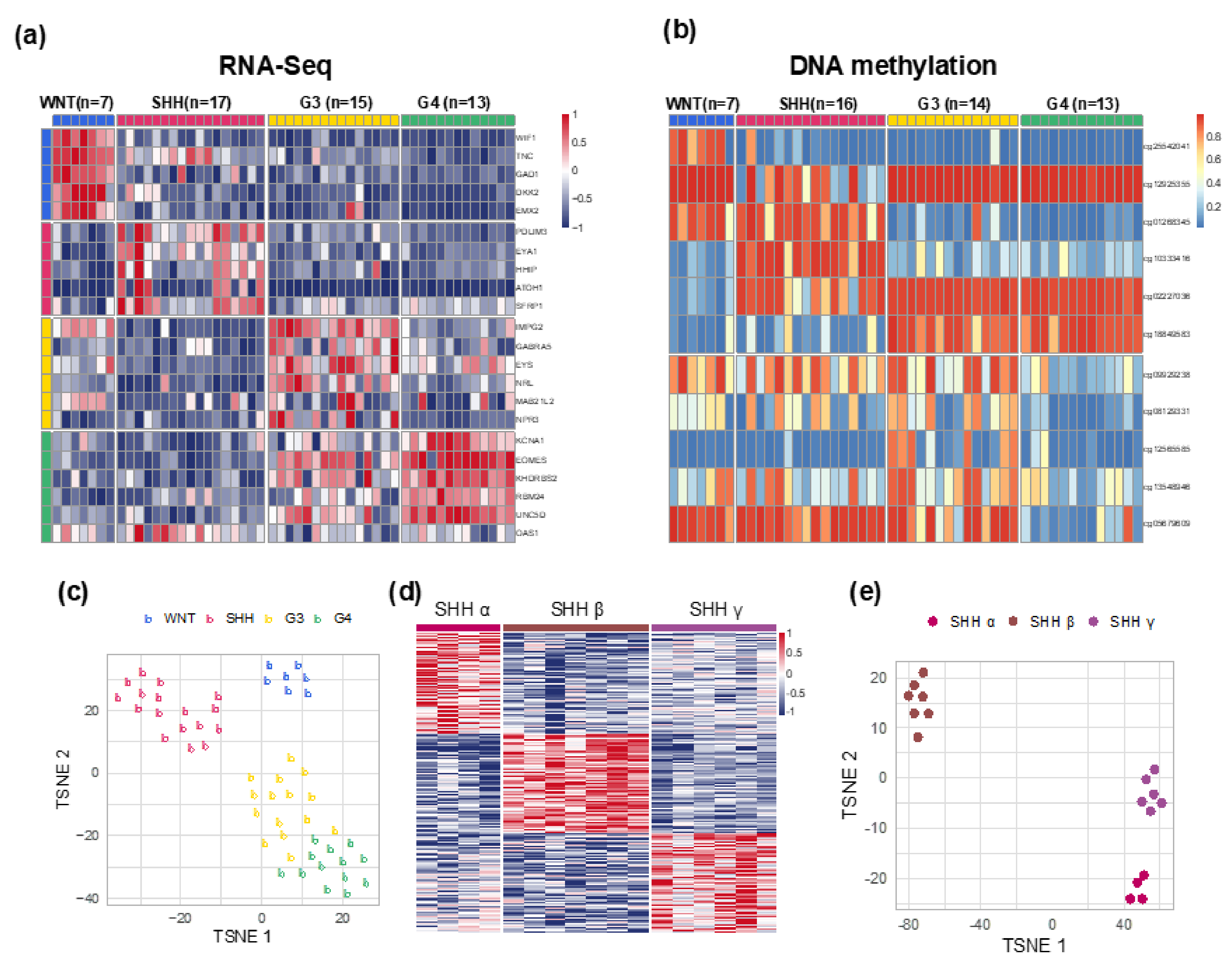
2.2. Molecular-Classification and Molecular-Clinical Correlation
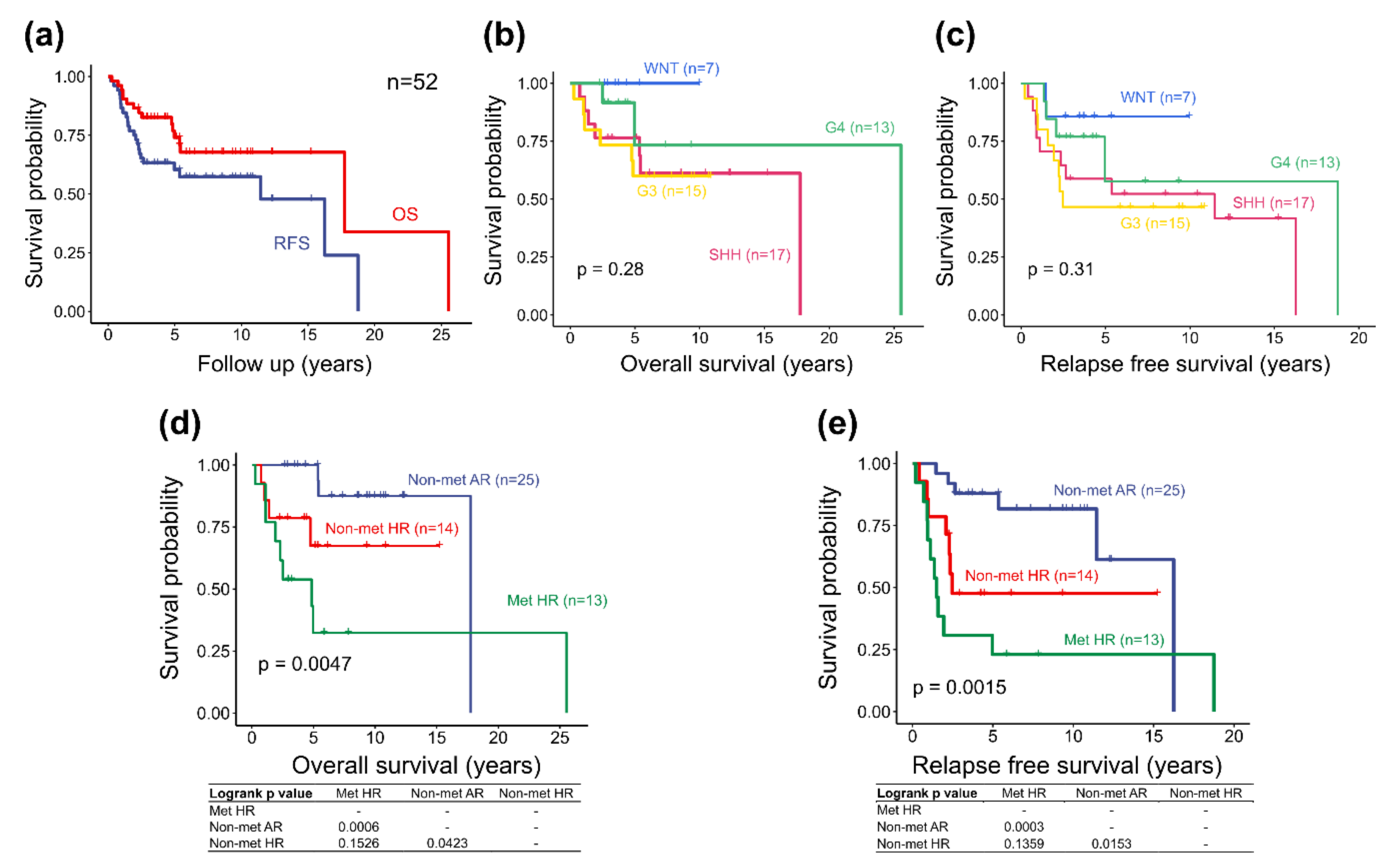
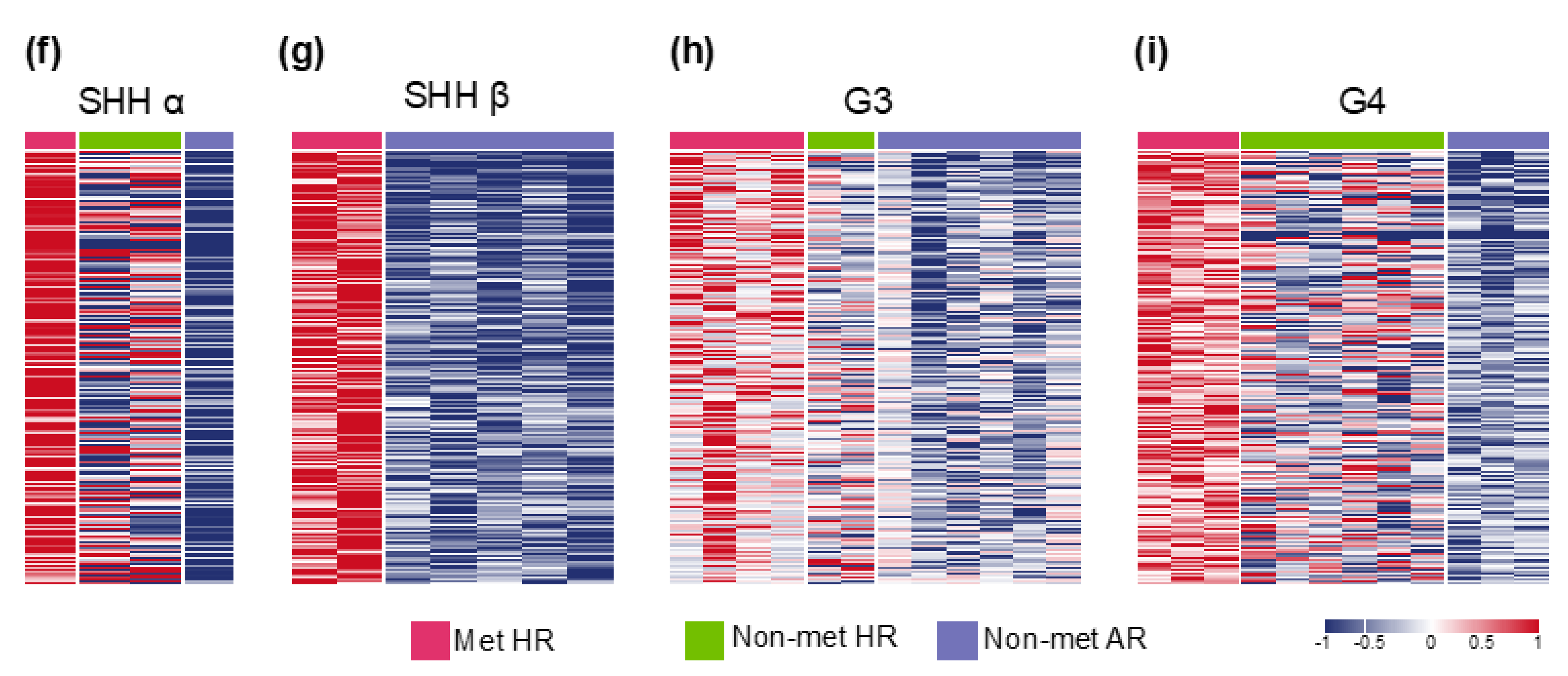
2.3. Molecular Subgroup-Based Clinical Risk Stratifications and Survivals
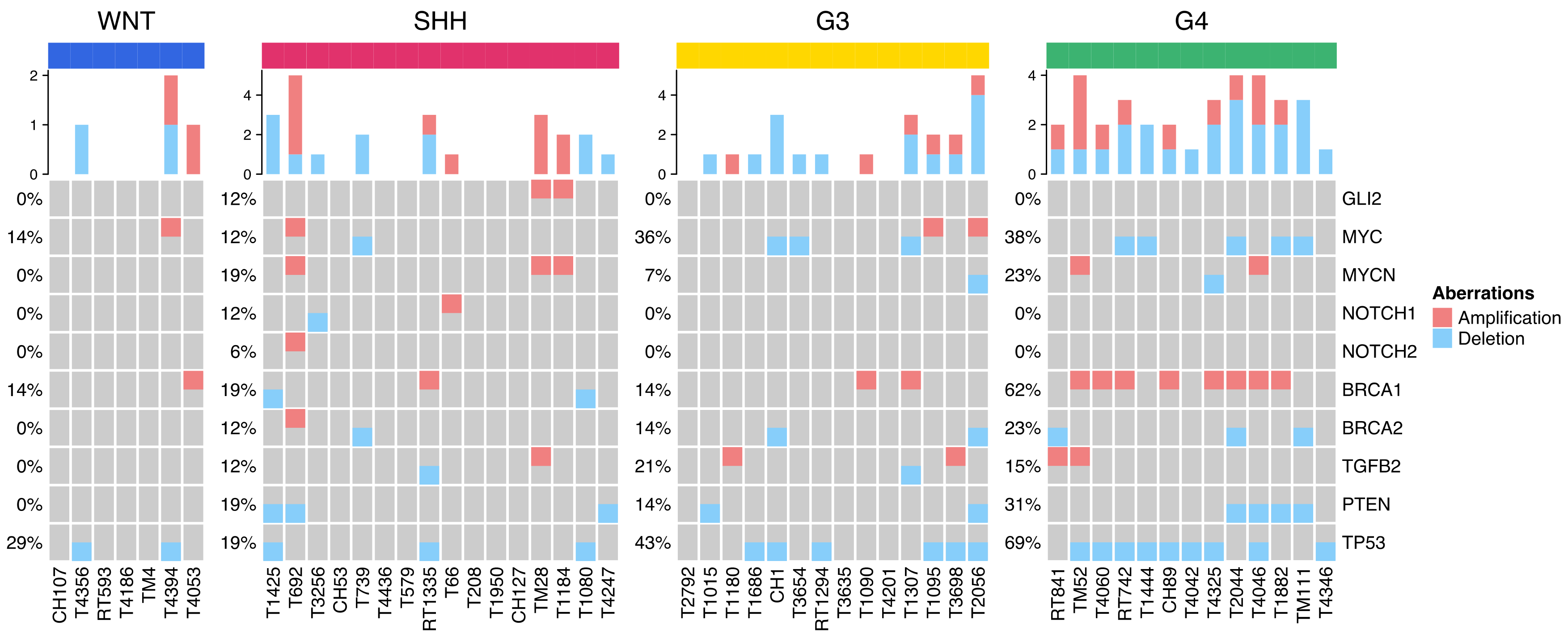
2.4. Distribution of Selected CNVs and Chromosomal Aberrations across MB Subgroups
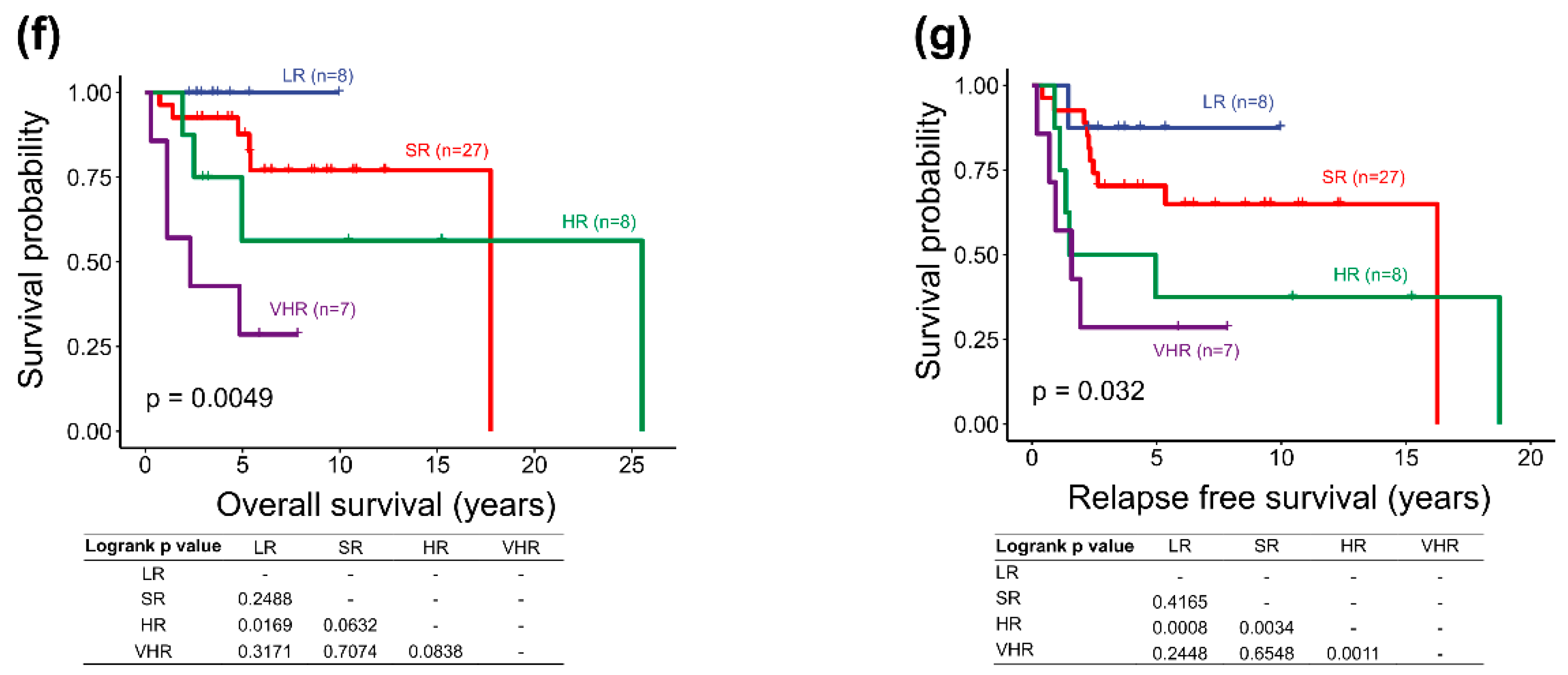
2.5. Survival Analysis of the Adjusted Heidelberg Scheme
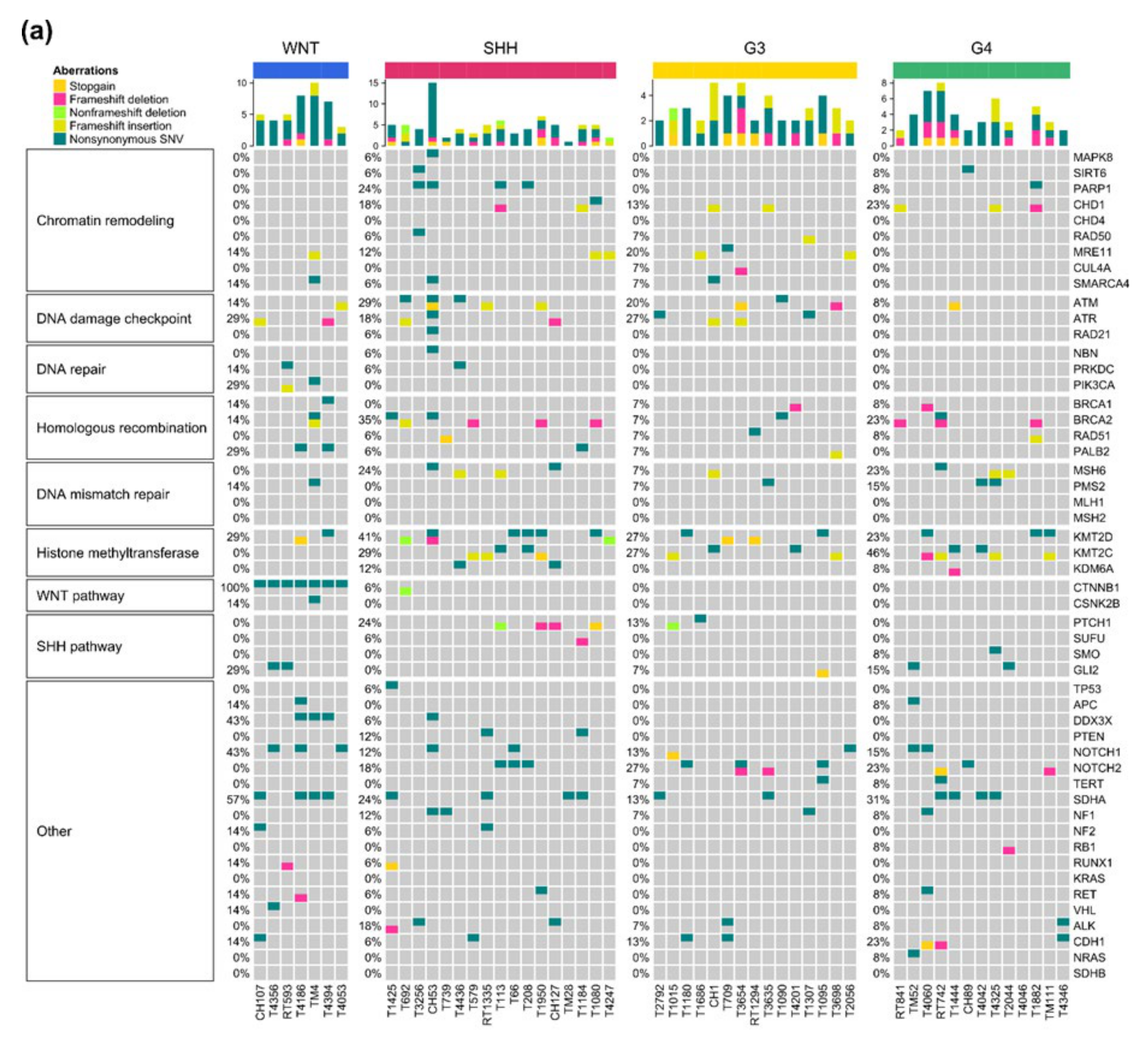
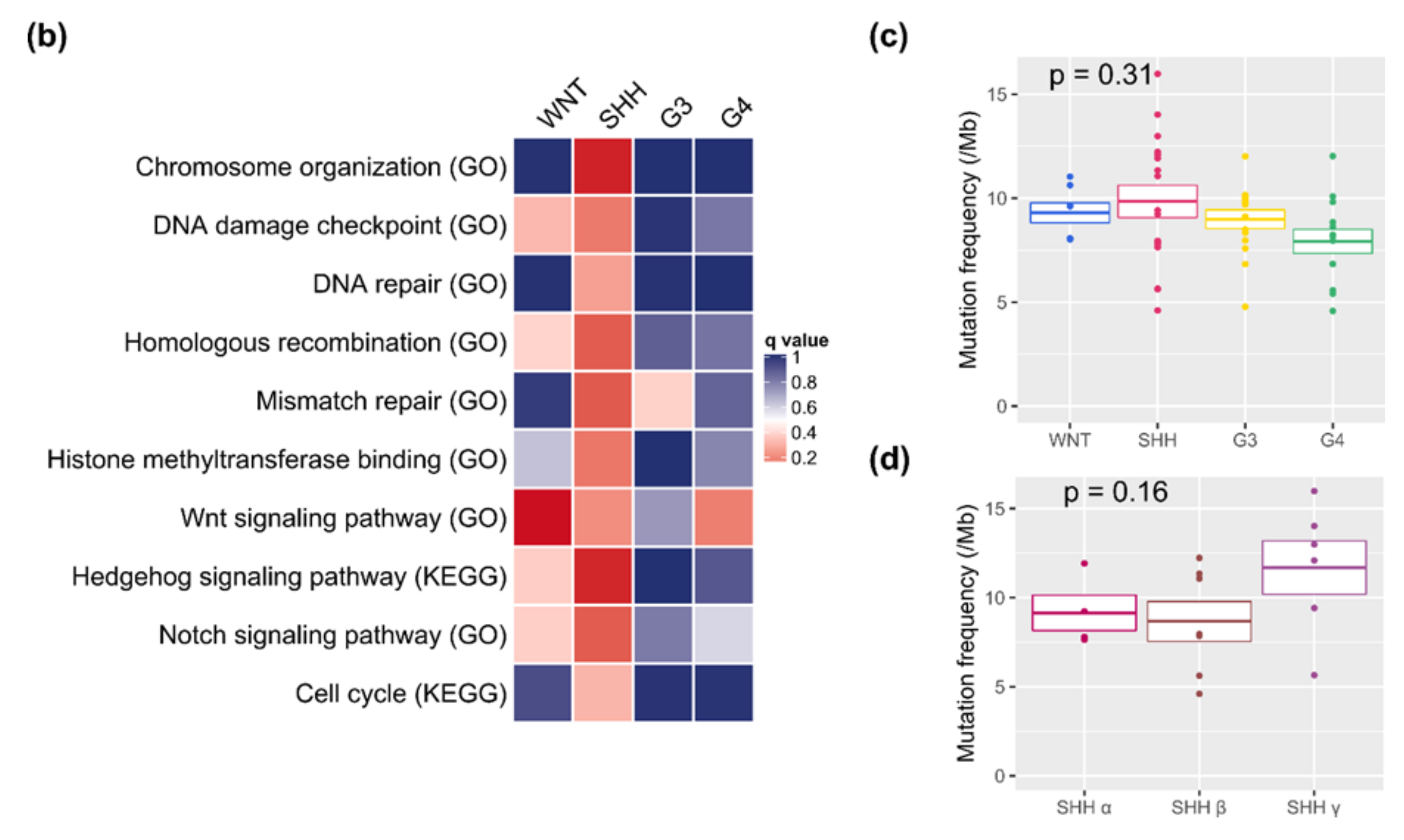
2.6. Somatic Mutations and Their Subgroup Distribution
2.7. Potential Association of Genetic Predisposition in Patients
3. Discussion
3.1. Potential Therapeutic Targeting from Mutations of the DDR
3.2. Finding Clues to Genetic Predisposition
4. Materials and Methods
4.1. Patient Cohort
4.2. Retrieval of Clinical Data
4.3. Molecular Diagnosis
4.4. Molecular Subgroup-Based Clinical Risk Stratification
4.5. The Adjusted Heidelberg Risk Stratification Scheme
4.6. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugieres, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2012, 19, 1541–1544. [Google Scholar] [CrossRef]
- Zeltzer, P.M.; Boyett, J.M.; Finlay, J.L.; Albright, A.L.; Rorke, L.B.; Milstein, J.M.; Allen, J.C.; Stevens, K.R.; Stanley, P.; Li, H.; et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: Conclusions from the Children’s Cancer Group 921 randomized phase III study. J. Clin. Oncol. 1999, 17, 832–845. [Google Scholar] [CrossRef]
- McManamy, C.S.; Pears, J.; Weston, C.L.; Hanzely, Z.; Ironside, J.W.; Taylor, R.E.; Grundy, R.G.; Clifford, S.C.; Ellison, D.W.; Clinical Brain Tumour, G. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Hielscher, T.; Bouffet, E.; Remke, M.; Luu, B.; Gururangan, S.; McLendon, R.E.; Bigner, D.D.; Lipp, E.S.; Perreault, S.; et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016, 17, 484–495. [Google Scholar] [CrossRef]
- Wong, T.T.; Liu, Y.L.; Ho, D.M.; Chang, K.P.; Liang, M.L.; Chen, H.H.; Lee, Y.Y.; Chang, F.C.; Lin, S.C.; Hsu, T.R.; et al. Factors affecting survival of medulloblastoma in children: The changing concept of management. Childs Nerv. Syst. 2015, 31, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Doz, F.; Demaille, M.C.; Tron, P.; Roche, H.; Plantaz, D.; Thyss, A.; Stephan, J.L.; Lejars, O.; Sariban, E.; et al. Improving survival in recurrent medulloblastoma: Earlier detection, better treatment or still an impasse? Br. J. Cancer 1998, 77, 1321–1326. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Robbins, C.J.; Bou-Dargham, M.J.; Sanchez, K.; Rosen, M.C.; Sang, Q.A. Decoding Somatic Driver Gene Mutations and Affected Signaling Pathways in Human Medulloblastoma Subgroups. J. Cancer 2018, 9, 4596–4610. [Google Scholar] [CrossRef]
- Coudray, A.; Battenhouse, A.M.; Bucher, P.; Iyer, V.R. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. PeerJ 2018, 6, e5362. [Google Scholar] [CrossRef]
- Wolff, A.; Perera-Bel, J.; Schildhaus, H.U.; Homayounfar, K.; Schatlo, B.; Bleckmann, A.; Beissbarth, T. Using RNA-Seq Data for the Detection of a Panel of Clinically Relevant Mutations. Stud. Health Technol. Inform. 2018, 253, 217–221. [Google Scholar] [PubMed]
- Piskol, R.; Ramaswami, G.; Li, J.B. Reliable identification of genomic variants from RNA-seq data. Am. J. Hum. Genet. 2013, 93, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, N.G.; Hansford, J.R.; McGlade, J.P.; Alvaro, F.; Ashley, D.M.; Bailey, S.; Baker, D.L.; Bourdeaut, F.; Cho, Y.J.; Clay, M.; et al. Medulloblastoma Down Under 2013: A report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol. 2014, 127, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.M.; Hawes, S.M.; Kees, U.R.; Gottardo, N.G.; Dallas, P.B. Gene expression analyses of the spatio-temporal relationships of human medulloblastoma subgroups during early human neurogenesis. PloS ONE 2014, 9, e112909. [Google Scholar] [CrossRef]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Yu, B.; O’Toole, S.A.; Trent, R.J. Somatic DNA mutation analysis in targeted therapy of solid tumours. Transl. Pediatr. 2015, 4, 125–138. [Google Scholar] [CrossRef]
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66–67, 24–29. [Google Scholar] [CrossRef]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, I.; Ackermann, A.; Duding, T.; Graband, A.; Filmann, N.; Plotz, G.; Zeuzem, S.; Brieger, A. Loss of MLH1 sensitizes colon cancer cells to DNA-PKcs inhibitor KU60648. Mol. Carcinog. 2017, 56, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; McCabe, N.; Mullarkey, M.; Cummins, R.; Burgess, D.J.; Nakabeppu, Y.; Oka, S.; Kay, E.; Lord, C.J.; Ashworth, A. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell 2010, 17, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Koi, M.; Balaguer, F.; Boland, C.R.; Goel, A. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J. Biol. Chem. 2011, 286, 12157–12165. [Google Scholar] [CrossRef]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J. Med. Genet. 2014, 51, 71–75. [Google Scholar] [CrossRef]
- Bourdeaut, F.; Delattre, O. Genetic predisposition to medulloblastomas: Just follow the tumour genome. Lancet Oncol. 2018, 19, 722–723. [Google Scholar] [CrossRef]
- Remke, M.; Ramaswamy, V.; Peacock, J.; Shih, D.J.; Koelsche, C.; Northcott, P.A.; Hill, N.; Cavalli, F.M.; Kool, M.; Wang, X.; et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013, 126, 917–929. [Google Scholar] [CrossRef]
- Vijapura, C.; Saad Aldin, E.; Capizzano, A.A.; Policeni, B.; Sato, Y.; Moritani, T. Genetic Syndromes Associated with Central Nervous System Tumors. Radiographics 2017, 37, 258–280. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, E.M.; Eppolito, A.; Henson, J.W. Genetic counseling and tumor predisposition in neuro-oncology practice. Neurooncol. Pract. 2016, 3, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, K.; Gururangan, S. Molecular variants and mutations in medulloblastoma. Pharmgenomics Pers. Med. 2014, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Farndon, P.A. Nevoid Basal Cell Carcinoma Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Smith, M.J.; Beetz, C.; Williams, S.G.; Bhaskar, S.S.; O’Sullivan, J.; Anderson, B.; Daly, S.B.; Urquhart, J.E.; Bholah, Z.; Oudit, D.; et al. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J. Clin. Oncol. 2014, 32, 4155–4161. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010, 95, 699–701. [Google Scholar] [CrossRef]
- Varley, J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum. Mutat. 2003, 21, 313–320. [Google Scholar] [CrossRef]
- Villani, A.; Tabori, U.; Schiffman, J.; Shlien, A.; Beyene, J.; Druker, H.; Novokmet, A.; Finlay, J.; Malkin, D. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study. Lancet Oncol. 2011, 12, 559–567. [Google Scholar] [CrossRef]
- De Queiroz Rossanese, L.B.; De Lima Marson, F.A.; Ribeiro, J.D.; Coy, C.S.; Bertuzzo, C.S. APC germline mutations in families with familial adenomatous polyposis. Oncol. Rep. 2013, 30, 2081–2088. [Google Scholar] [CrossRef]
- Hobert, J.A.; Eng, C. PTEN hamartoma tumor syndrome: An overview. Genet. Med. 2009, 11, 687–694. [Google Scholar] [CrossRef]
- Tabori, U.; Baskin, B.; Shago, M.; Alon, N.; Taylor, M.D.; Ray, P.N.; Bouffet, E.; Malkin, D.; Hawkins, C. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J. Clin. Oncol. 2010, 28, 1345–1350. [Google Scholar] [CrossRef]
- Scott, R.H.; Mansour, S.; Pritchard-Jones, K.; Kumar, D.; MacSweeney, F.; Rahman, N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat. Clin. Pract. Oncol. 2007, 4, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Rankin, S.L.; Larson, J.D.; Zhu, X.; Chow, L.M.; Qu, C.; Zhang, J.; Ellison, D.W.; Baker, S.J. PTEN Signaling in the Postnatal Perivascular Progenitor Niche Drives Medulloblastoma Formation. Cancer Res. 2017, 77, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, B.K.; Stenzel, T.T.; McLendon, R.E.; Parsons, R.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Bigner, S.H. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997, 57, 4187–4190. [Google Scholar] [PubMed]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Evans, D.G.; Farndon, P.A.; Burnell, L.D.; Gattamaneni, H.R.; Birch, J.M. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br. J. Cancer 1991, 64, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Teot, L.A.; Kozakewich, H.; Werger, A.M.; Davies, K.J.; Fletcher, C.D.; Grier, H.E.; Rodriguez-Galindo, C.; Wagers, A.J. Myogenic tumors in nevoid Basal cell carcinoma syndrome. J. Pediatr. Hematol. Oncol. 2015, 37, 147–149. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Chang, C.H.; Housepian, E.M.; Herbert, C., Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 1969, 93, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Taylor, M.D. Medulloblastoma: From Myth to Molecular. J. Clin. Oncol. 2017, 35, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, e1521. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, e367. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Remke, M.; Cho, Y.J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012, 123, 615–626. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Gomez, S.; Garrido-Garcia, A.; Garcia-Gerique, L.; Lemos, I.; Sunol, M.; de Torres, C.; Kulis, M.; Perez-Jaume, S.; Carcaboso, A.M.; Luu, B.; et al. A Novel Method for Rapid Molecular Subgrouping of Medulloblastoma. Clin. Cancer Res. 2018, 24, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Remke, M.; Kool, M.; Pietsch, T.; Northcott, P.A.; Fischer, R.; Cavalli, F.M.; Ramaswamy, V.; Zapatka, M.; Reifenberger, G.; et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013, 125, 913–916. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Subgroup Assignment | ||||||
|---|---|---|---|---|---|---|
| N = 52 | WNT n = 7 (13.5%) | SHH n = 17 (32.7%) | Group 3 n = 15 (28.8%) | Group 4 n = 13 (25%) | ||
| SHH α, n = 4 | SHH β, n = 7 | SHH γ, n = 6 | ||||
| Frozen tissue for molecular profiling | ||||||
| Primary tumor | 7 | 4 | 7 | 6 | 12 | 12 |
| Recurrent tumor | 0 | 0 | 0 | 0 | 3 | 1 |
| Age (median, range) at diagnosis (years) | ||||||
| 7.2 (0.3–18.2) | 8.5 (3.1–11.4) | 4.0 (0.3–14.3) | 6.0 (1.6–18.2) | 10.1 (5.1–15.4) | ||
| 7.4 (2.8–12.6) | 4.2 (3.7–14.3) | 1.7 (0.3–5.8) | ||||
| ≤3 y (n = 8, 15.4%) | 0 | 1 (25%) | 0 | 5 (83.3%) | 2 (13.3%) | 0 |
| >3 y (n = 44, 84.6%) | 7 (100%) | 3 (75%) | 7 (100%) | 1 (16.7%) | 13 (86.7%) | 13 (100%) |
| Sex | ||||||
| Male, n = 28 (53.8%) | 1 | 2 | 4 | 4 | 9 | 8 |
| Female, n = 24 (46.2%) | 6 | 2 | 3 | 2 | 6 | 5 |
| Male/female ratio (1.2/1) | 0.2/1 | 1.4/1 | 1.5/1 | 1.6/1 | ||
| 1/1 | 1.3/1 | 2/1 | ||||
| Metastasis stage at diagnosis (M0-1, M2-3), number of cases (percentage) | ||||||
| M0-1, n = 39 (75%) | 7 (100%) | 3 (75%) | 5 (71.4%) | 6 (100%) | 9 (60%) | 9 (69.2%) |
| M2-3, n = 13 (25%) | 0 | 3 (17.6%) | 6 (40%) | 4 (30.8%) | ||
| 1 (25%) | 2 (28.6%) | 0 | ||||
| Extent of resection (NTR-TR, residue tumor < 1.5 cm2; STR, residue tumor ≥ 1.5 cm2), number of cases (percentage) | ||||||
| NTR-TR (n = 29, 55.8%) | 5 (71.4%) | 1 (25%) | 6 (85.7%) | 3 (50%) | 9 (60%) | 5 (38.5%) |
| STR (n = 23, 44.2%) | 2 (28.6%) | 3 (75%) | 1 (14.3%) | 3 (50%) | 6 (40%) | 8 (61.5%) |
| Pathology variant, number of cases (percentage) and median age (years) | ||||||
| Classic, n = 27 (51.9%), 6.3 | 5 (71.4%), | 1 (25%) | 6 (85.7%) | 2 (33.3%) | 7 (46.7%) | 6 (46.2%) |
| DNMB, n = 8 (15.4%), 2.8 | 1 (14.3%) | 2 (50%) | 1 (14.3%) | 3 (50%) | 0 | 1 (7.7%) |
| MBEN, n = 1 (1.9%), 1.0 | 0 | 0 | 0 | 1 (16.7%) | 0 | 0 |
| LCA, n = 15 (28.8%), 8.5 | 1 (14.3%) | 1 (25%) | 0 | 0 | 7 (46.7%) | 6 (46.2%) |
| MMMB, n = 1 (1.9%), 4.3 | 0 | 0 | 0 | 0 | 1 (6.7%) | 0 |
| Molecular subgroup-based clinical risk stratification *, number of cases | ||||||
| Non-met (M0-1) AR, n = 25 | 7 | 1 | 5 | 3 | 6 | 3 |
| Non-met (M0-1) HR, n = 14 | 0 | 2 | 0 | 3 | 3 | 6 |
| Met (M2-3) HR, n = 13 | 0 | 1 | 2 | 0 | 6 | 4 |
| Treatment strategy, number of cases (percentage) | ||||||
| CMT alone, n = 3 (5.8%) | 0 | 0 | 0 | 3 | 0 | 0 |
| RT alone, n = 4 (7.7%) | 0 | 0 | 0 | 1 | 2 | 1 |
| RT + CMT, n = 45 (86.5%) | 7 | 4 | 7 | 2 | 13 | 12 |
| Recurrence, number of cases (percentage) | ||||||
| Recurrence, n = 21 (40.4%) | 1 (14.3%) | 8 (47.1%) | 8 (53.3%) | 4 (30.8%) | ||
| 1 (25%) | 4 (57.1%) | 3 (50%) | ||||
| Time from diagnosis to the first recurrence (years) | ||||||
| Median time (range) | 1.5 | 0.7 | 1.9 (0.9–16.3) | 0.9 (0.4–2.3) | 1.8 (0.2–2.5) | 1.8 (1.4–19.5) |
| Median follow-up time (range) (years) | ||||||
| 5.2 (0.3–25.6) | 3.7(2.7–10.0) | 5.8 (1.1–15.3) | 12.3 (1.9–17.7) | 4.0 (0.8–10.5) | 6.5 (0.3–10.9). | 4.3 (1.7–25.6) |
| Survivals of molecular subgroup (percentage) | ||||||
| 5-year OS rate: 74.1% | 100% | 76.5% | 60.0% | 72.7% | ||
| 5-year RFS rate: 60.4% | 85.7% | 58.8% | 46.7% | 61.4% | ||
| Subgroup/Survival Rate | Low-Risk (>90% Survival) | Standard-Risk (75–90% Survival) | High-Risk (50–75% Survival) | Very High-Risk (<50% Survival) | Undetermined |
|---|---|---|---|---|---|
| WNT | <16 y, Non-metastatic (M0-1) | Metastatic (M2-3) | |||
| SHH | All of the following: TP53 WT (somatic), No MYCN amplification, Non-metastasis (M0-1) | One or both: MYCN amplification, Metastatic (M2-3) | TP53 mutation (metastatic or non-metastatic) | No data of MYCN amplification | |
| Group 3 | All of the following: No MYC amplification, Non-metastatic (M0-1) | Metastatic (M2-3) | No data of MYC amplification, or Non-metastatic with MYC amplification | ||
| Group 4 | All of the following: Non-metastatic (M0-1), Chr. 11 loss | All of the following: Non-metastatic (M0-1), No Chr. 11 loss | Metastatic (M2-3) |
| Pathway | Gene | WNT, n (%) | SHH, n (%) | G3, n (%) | G4, n (%) |
|---|---|---|---|---|---|
| Chromatin remodeling | MAPK8 (1.9%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 0 (0%) |
| SIRT6 (3.8%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 1 (1.9%) | |
| PARP1 (9.6%) | 0 (0%) | 4 (7.7%) | 0 (0%) | 1 (1.9%) | |
| CHD1 (15.4%) | 0 (0%) | 3 (5.8%) | 2 (3.8%) | 3 (5.8%) | |
| CHD4 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| RAD50 (3.8%) | 0 (0%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | |
| MRE11 (11.5%) | 1 (1.9%) | 2 (3.8%) | 3 (5.8%) | 0 (0%) | |
| CUL4A (1.9%) | 0 (0%) | 0 (0%) | 1 (1.9%) | 0 (0%) | |
| SMARCA4 (5.8%) | 1 (1.9%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | |
| DNA damage checkpoint | ATM (19.2%) | 1 (1.9%) | 5 (9.6%) | 3 (5.8%) | 1 (1.9%) |
| ATR (17.3%) | 2 (3.8%) | 3 (5.8%) | 4 (7.7%) | 0 (0%) | |
| RAD21 (1.9%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| DNA repair | NBN (1.9%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 0 (0%) |
| PRKDC (3.8%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| PIK3CA (3.8%) | 2 (3.8%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| Homologous recombination | BRCA1 (5.8%) | 1 (1.9%) | 0 (0%) | 1 (1.9%) | 1 (1.9%) |
| BRCA2 (21.2%) | 1 (1.9%) | 6 (11.5%) | 1 (1.9%) | 3 (5.8%) | |
| RAD51 (5.8%) | 0 (0%) | 1 (1.9%) | 1 (1.9%) | 1 (1.9%) | |
| PALB2 (7.7%) | 2 (3.8%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | |
| DNA mismatch repair | MSH6 (15.4%) | 0 (0%) | 4 (7.7%) | 1 (1.9%) | 3 (5.8%) |
| PMS2 (7.7%) | 1 (1.9%) | 0 (0%) | 1 (1.9%) | 2 (3.8%) | |
| MLH1 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| MSH2 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| Histone methyltransferase | KMT2D (30.8%) | 2 (3.8%) | 7 (13.5%) | 4 (7.7%) | 3 (5.8%) |
| KMT2C (28.8%) | 0 (0%) | 5 (9.6%) | 4 (7.7%) | 6 (11.5%) | |
| KDM6A (5.8%) | 0 (0%) | 2 (3.8%) | 0 (0%) | 1 (1.9%) | |
| WNT pathway | CTNNB1 (15.4%) | 7 (13.5%) | 1 (1.9%) | 0 (0%) | 0 (0%) |
| CSNK2B (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| SHH pathway | PTCH1 (11.5%) | 0 (0%) | 4 (7.7%) | 2 (3.8%) | 0 (0%) |
| SUFU (1.9%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| SMO (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.9%) | |
| GLI2 (9.6%) | 2 (3.8%) | 0 (0%) | 1 (1.9%) | 2 (3.8%) | |
| Other | TP53 (1.9%) | 0 (0%) | 1 (1.9%) | 0 (0%) | 0 (0%) |
| APC (3.8%) | 1 (1.9%) | 0 (0%) | 0 (0%) | 1 (1.9%) | |
| DDX3X (7.7%) | 3 (5.8%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| PTEN (3.8%) | 0 (0%) | 2 (3.8%) | 0 (0%) | 0 (0%) | |
| NOTCH1 (17.3%) | 3 (5.8%) | 2 (3.8%) | 2 (3.8%) | 2 (3.8%) | |
| NOTCH2 (19.2%) | 0 (0%) | 3 (5.8%) | 4 (7.7%) | 3 (5.8%) | |
| TERT (3.8%) | 0 (0%) | 0 (0%) | 1 (1.9%) | 1 (1.9%) | |
| SDHA (26.9%) | 4 (7.7%) | 4 (7.7%) | 2 (3.8%) | 4 (7.7%) | |
| NF1 (7.7%) | 0 (0%) | 2 (3.8%) | 1 (1.9%) | 1 (1.9%) | |
| NF2 (3.8%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| RB1 (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.9%) | |
| RUNX1 (3.8%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | |
| KRAS (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| RET (5.8%) | 1 (1.9%) | 1 (1.9%) | 0 (0%) | 1 (1.9%) | |
| VHL (1.9%) | 1 (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| ALK (9.6%) | 0 (0%) | 3 (5.8%) | 1 (1.9%) | 1 (1.9%) | |
| CDH1 (13.5%) | 1 (1.9%) | 1 (1.9%) | 2 (3.8%) | 3 (5.8%) | |
| NRAS (1.9%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1.9%) | |
| SDHB (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Case No. | Age at Dx/Sex | Molecular Subtypes | Histological Phenotype | Metastatic Status at Dx | Relevant Clinical Findings | Driver Mutation | Associated Somatic Mutations | Potential Genetic Predisposition | Mutation Frequency (/Mb) † | Post-Resection Treatment | Outcome/Age at FU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8.5 y/F | SHH α | LCA | M3 | Early recurrence | TP53 | BRCA2, SDHA, RUNX1, ALK | TP53 germline mutation (Li-Fraumeni syndrome) § | 7.64 | RT (CSI + PFI) + CMT | Died at 9.6 y/o from recurrence |
| 2 | 6.3 y/F | SHH α | DNMB | M0-1 | Anaplastic astrocytoma at 8.1 y/o, T-lymphoblastic lymphoma at 11 y/o | MSH6 | PRPA1, SMARCA4, ATM, ATR, RAD21, KMT2D, NOTCH1 | Biallelic MSH6 germline mutation (CMMR-D) | 9.23 | RT (CSI + PFI) + CMT | Died at 11.6 y/o from progressing anaplastic astrocytoma |
| 3 | 3.6 y/F | SHH β ‡ | Classic | M0-1 | Multiple adenomatous hyperplasia and nodular goiter at 3.8 y/o | PTEN | ATM, KMT2C, SHHA, NF2 | PTEN germline mutation (PHTS) § | 5.62 | RT (CSI + PFI) | Died at 11.4 y/o from recurrence |
| 4 | 5.8 y/F | SHH γ | Classic | M0-1 | Parotid rhabdomyosarcoma at 4.3 y/o | PTCH1 | CHD1, MRE11, BRCA2, KMT2D | PTCH1 somatic mutation (Gorlin syndrome) | 5.65 | CMT | Alive at 14.3 y/o |
| 5 | 14.5 y/M | SHH β | Classic | M0-1 | Buccal myxofibrosarcoma at 17.2 y/o | TERT * | PARP1, KMT2, KMT2D, NOTCH2 | TERT promotor mutation * | 7.95 | RT (CSI + PFI) + CMT | Alive at 26 y/o |
| 6 | 2.2 y/M | SHH γ | DNMB | M0-1 | No | SUFU | PALB2, PTEN, SDHA | SUFU and PALB2 germline mutation (Gorlin syndrome) | 15.98 | RT (PFI) + CMT | Alive at 12.7 y/o |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.-S.; Ho, D.M.-T.; Jou, S.-T.; Yu, A.L.; Tran, H.M.; Liang, M.-L.; Chen, H.-H.; Lee, Y.-Y.; Chen, Y.-W.; Lin, S.-C.; et al. Molecular-Clinical Correlation in Pediatric Medulloblastoma: A Cohort Series Study of 52 Cases in Taiwan. Cancers 2020, 12, 653. https://doi.org/10.3390/cancers12030653
Wu K-S, Ho DM-T, Jou S-T, Yu AL, Tran HM, Liang M-L, Chen H-H, Lee Y-Y, Chen Y-W, Lin S-C, et al. Molecular-Clinical Correlation in Pediatric Medulloblastoma: A Cohort Series Study of 52 Cases in Taiwan. Cancers. 2020; 12(3):653. https://doi.org/10.3390/cancers12030653
Chicago/Turabian StyleWu, Kuo-Sheng, Donald Ming-Tak Ho, Shiann-Tarng Jou, Alice L. Yu, Huy Minh Tran, Muh-Lii Liang, Hsin-Hung Chen, Yi-Yen Lee, Yi-Wei Chen, Shih-Chieh Lin, and et al. 2020. "Molecular-Clinical Correlation in Pediatric Medulloblastoma: A Cohort Series Study of 52 Cases in Taiwan" Cancers 12, no. 3: 653. https://doi.org/10.3390/cancers12030653
APA StyleWu, K.-S., Ho, D. M.-T., Jou, S.-T., Yu, A. L., Tran, H. M., Liang, M.-L., Chen, H.-H., Lee, Y.-Y., Chen, Y.-W., Lin, S.-C., Chang, F.-C., Tsai, M.-L., Liu, Y.-L., Lee, H.-L., Hsieh, K. L.-C., Huang, W.-C., Sung, S.-Y., Chang, C.-C., Changou, C. A., ... Wong, T.-T. (2020). Molecular-Clinical Correlation in Pediatric Medulloblastoma: A Cohort Series Study of 52 Cases in Taiwan. Cancers, 12(3), 653. https://doi.org/10.3390/cancers12030653