Exon-4 Mutations in KRAS Affect MEK/ERK and PI3K/AKT Signaling in Human Multiple Myeloma Cell Lines

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
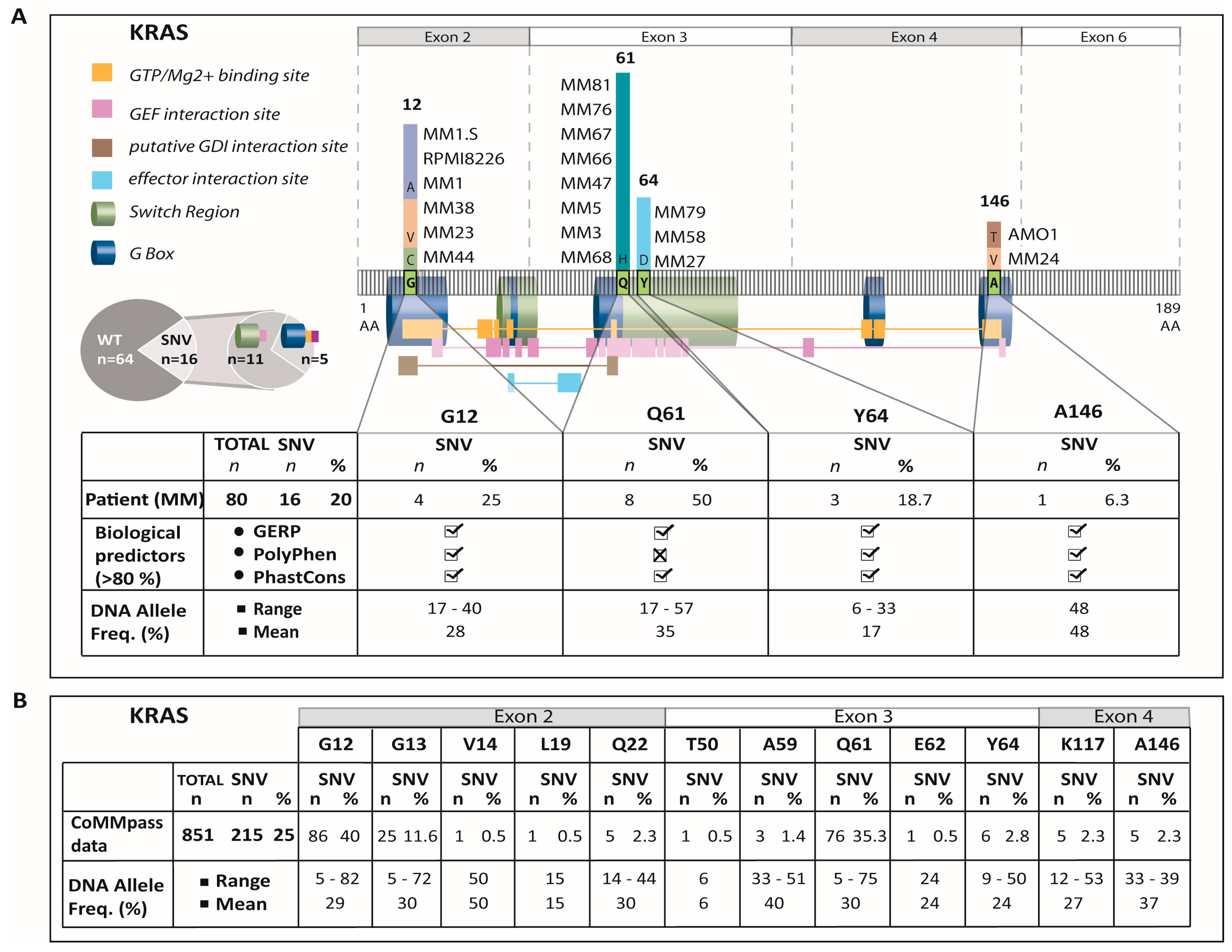
2.1. Sequencing, Filtering, and Validation
2.2. Exon-4 KRAS-Mutations Are Rare in the Current Study Cohort
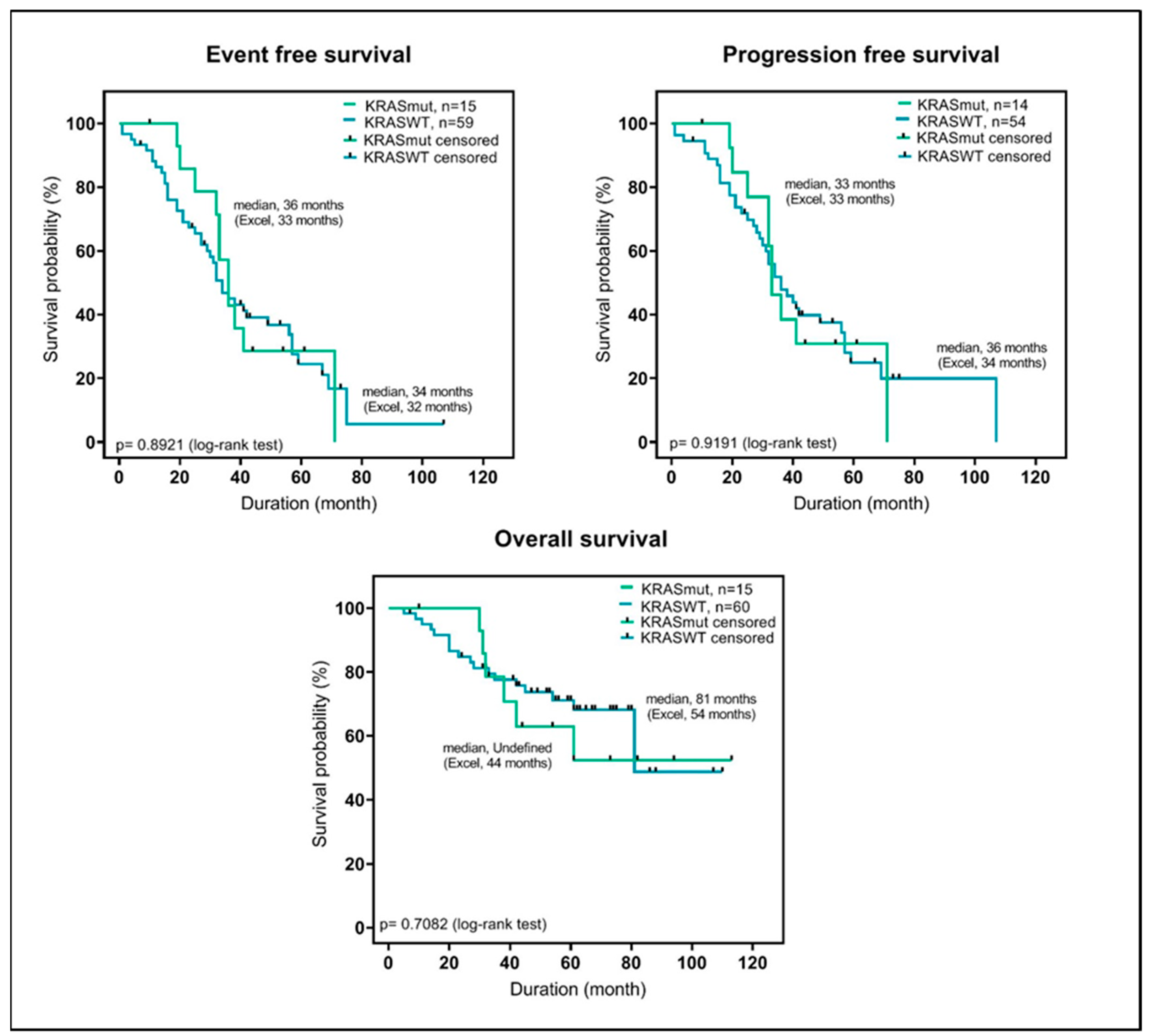
2.3. KRAS-Mutations at Diagnosis Have No Predictive Value in Patients Treated with VCD and High-Dose Chemotherapy
2.4. Exon-4 Mutation in AMO1 Cells is Accompanied by Increased CN-Stage and Gene Expression Levels
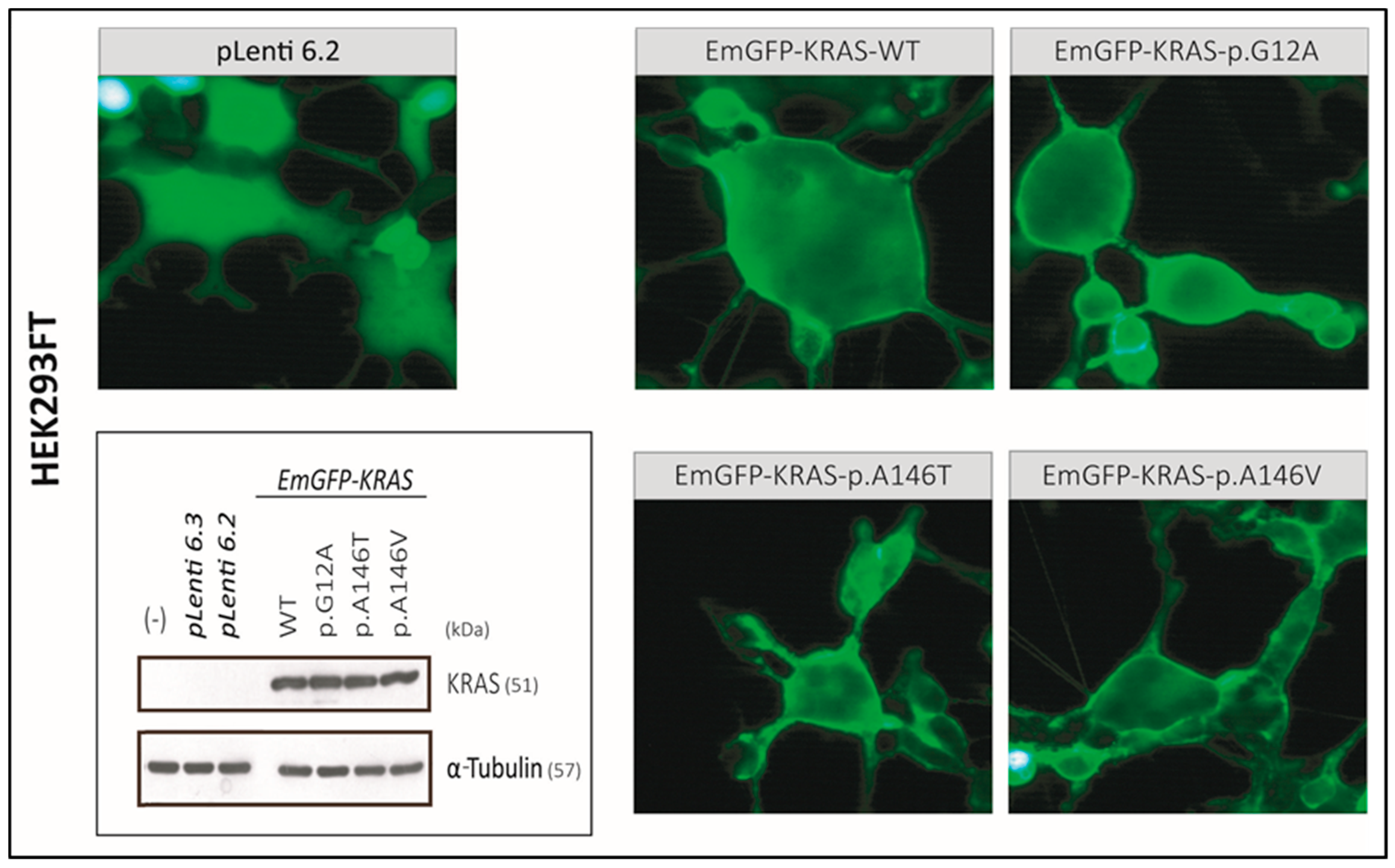
2.5. Exon-4 Mutations Do Not Appear to Influence the Cellular Localization of KRAS Protein
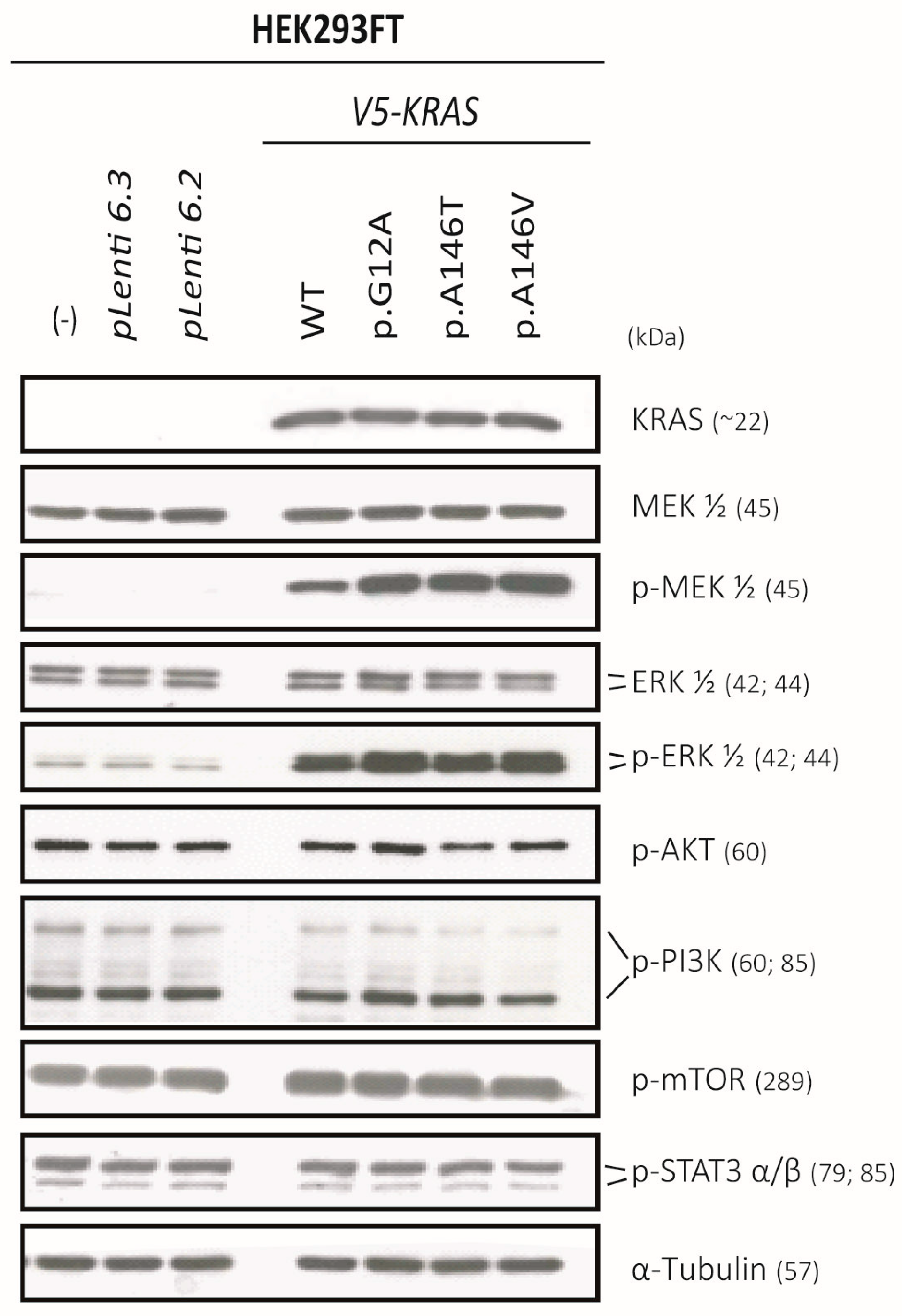
2.6. Exon-4 Mutations Specifically Activate MEK and ERK in HEK293 Cells
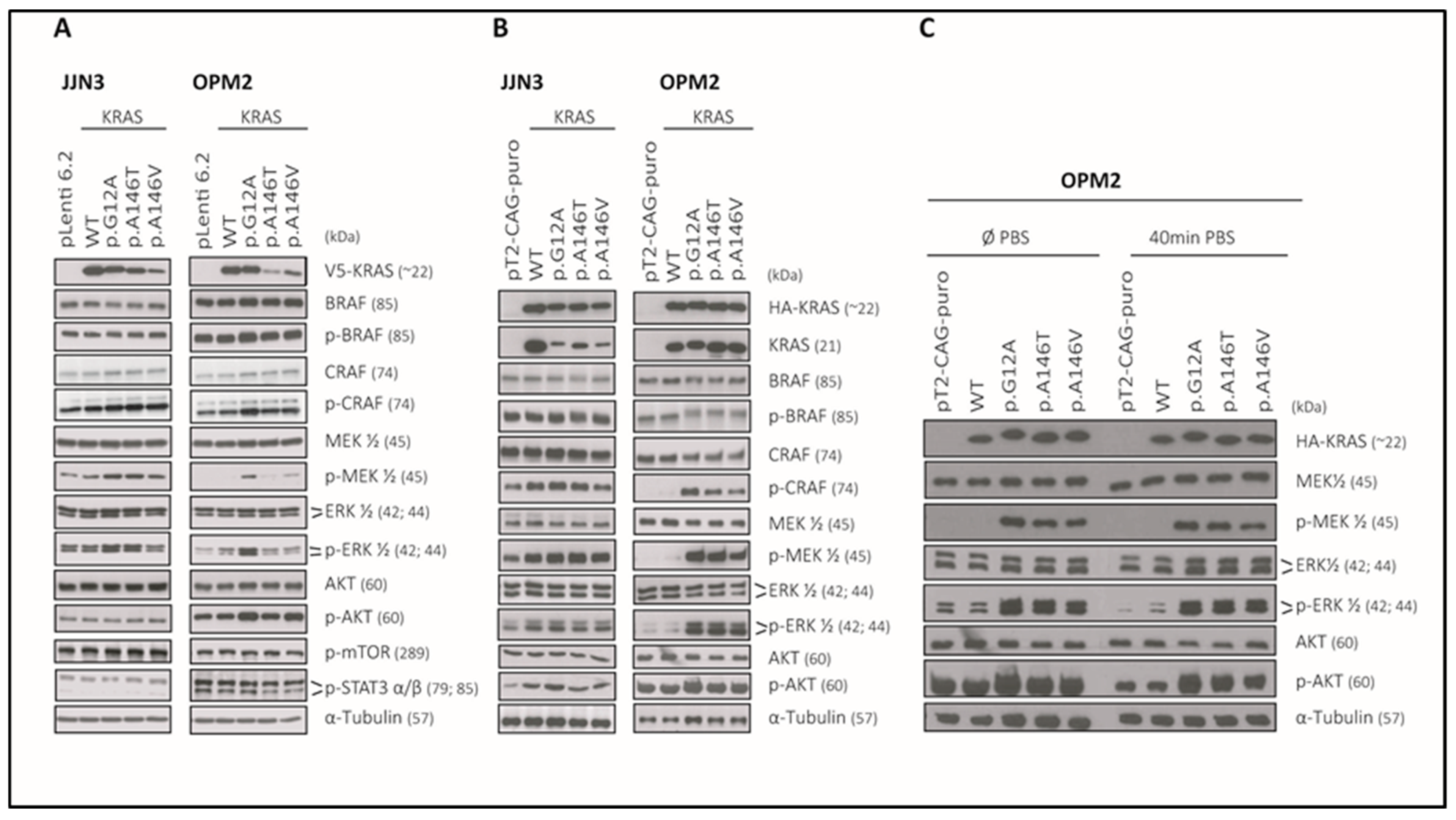
2.7. The KRAS-Mutants KRASp.G12A/p.A146T/p.A146V Specifically Activate MEK/ERK-Signaling in the KRASWT MM Cell Lines JJN3 and OPM2 and Can Sustain AKT Signalling in OPM2
3. Discussion
4. Materials and Methods
4.1. Patient Specimens and Human Cell Lines
4.2. Amplicon Generation and Sequencing
4.3. Sequencing Data Analysis and Technical Verification
4.4. Sanger Sequencing and High Resolution Meling (HRM)
4.5. Statistical Analysis
4.6. Generation of Transient and Stable KRAS-WT and KRAS-Mutant (p.G12A, p.A146T, p.A146V) Overexpression and of KRAS-Knockdown Cell Lines
4.7. Isolation of RNA and cDNA Synthesis
4.8. SNP 6.0 and HGU133 Plus 2.0 Microarray Analysis
4.9. Generation of Donor and Expression Vectors for Lentiviral Transduction
4.10. Virus Production in HEK293FT Cells
4.11. Viral Transduction of the MM Cell Lines OPM2 and JJN3
4.12. Generation of Stable Overexpression Cell Lines Using the Sleeping Beauty System
4.13. Transfection of HEK293FT Cells with Lipofectamine 2000
4.14. FACS Analysis
4.15. SDS PAGE and Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCormick, F. Progress in targeting RAS with small molecule drugs. Biochem. J. 2019, 476, 365–374. [Google Scholar] [CrossRef]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.V.; Oken, M.M.; Kyle, R.A.; Henderson, K.J.; Van Wier, S.; Greipp, P.; et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Jancik, S.; Drabek, J.; Radzioch, D.; Hajduch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Repasky, G.A.; Chenette, E.J.; Der, C.J. Renewing the conspiracy theory debate: Does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004, 14, 639–647. [Google Scholar] [CrossRef]
- Vakiani, E.; Solit, D.B. KRAS and BRAF: Drug targets and predictive biomarkers. J. Pathol. 2011, 223, 219–229. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Steinbrunn, T.; Stuhmer, T.; Gattenlohner, S.; Rosenwald, A.; Mottok, A.; Unzicker, C.; Einsele, H.; Chatterjee, M.; Bargou, R.C. Mutated RAS and constitutively activated Akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood 2011, 117, 1998–2004. [Google Scholar] [CrossRef]
- Leich, E.; Steinbrunn, T. RAS mutations—For better or for worse in multiple myeloma? Leuk. Lymphoma. 2016, 57, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Corradini, P.; Ladetto, M.; Voena, C.; Palumbo, A.; Inghirami, G.; Knowles, D.M.; Boccadoro, M.; Pileri, A. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias. Blood 1993, 81, 2708–2713. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, N.; Biersack, H.; Czerwinska, A.C.; Schemme, J.; Hardel, T.T.; Bernard, V.; Rades, D.; Lehnert, H.; Luley, K.B.; Thorns, C. Favorable prognostic impact of RAS mutation status in multiple myeloma treated with high-dose melphalan and autologous stem cell support in the era of novel agents: A single center perspective. Leuk. Lymphoma 2016, 57, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Way, G.P.; Barwick, B.G.; Mariano, M.C.; Marcoulis, M.; Ferguson, I.D.; Driessen, C.; Boise, L.H.; Greene, C.S.; Wiita, A.P. Integrated phosphoproteomics and transcriptional classifiers reveal hidden RAS signaling dynamics in multiple myeloma. Blood Adv. 2019, 3, 3214–3227. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Leong, T.; Quam, L.; Billadeau, D.; Kay, N.E.; Greipp, P.; Kyle, R.A.; Oken, M.M.; Van Ness, B. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: Analysis of the Eastern Cooperative Oncology Group Phase III Trial. Blood 1996, 88, 2699–2706. [Google Scholar] [CrossRef]
- Neri, A.; Murphy, J.P.; Cro, L.; Ferrero, D.; Tarella, C.; Baldini, L.; Dalla-Favera, R. Ras oncogene mutation in multiple myeloma. J. Exp. Med. 1989, 170, 1715–1725. [Google Scholar] [CrossRef]
- Rasmussen, T.; Kuehl, M.; Lodahl, M.; Johnsen, H.E.; Dahl, I.M. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood 2005, 105, 317–323. [Google Scholar] [CrossRef]
- Smith, D.; Armenteros, E.; Percy, L.; Kumar, M.; Lach, A.; Herledan, G.; Stubbs, M.; Downward, J.; Yong, K. RAS mutation status and bortezomib therapy for relapsed multiple myeloma. Br. J. Haematol. 2015, 169, 905–908. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Hulkki, S.; Potter, N.E.; Johnson, D.C.; Fenwick, K.; Kozarewa, I.; Gonzalez, D.; Lord, C.J.; et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012, 120, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Edkins, S.; O’Meara, S.; Parker, A.; Stevens, C.; Reis, M.; Jones, S.; Greenman, C.; Davies, H.; Dalgliesh, G.; Forbes, S.; et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 2006, 5, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Janakiraman, M.; Vakiani, E.; Zeng, Z.; Pratilas, C.A.; Taylor, B.S.; Chitale, D.; Halilovic, E.; Wilson, M.; Huberman, K.; Ricarte Filho, J.C.; et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010, 70, 5901–5911. [Google Scholar] [CrossRef]
- Multiple Myeloma Research Foundation Personalized Medicine Initiatives. CoMMpass Dataset, 2014 ed. 2014. Available online: https://research.themmrf.org (accessed on 16 February 2020).
- Leich, E.; Weissbach, S.; Klein, H.U.; Grieb, T.; Pischimarov, J.; Stuhmer, T.; Chatterjee, M.; Steinbrunn, T.; Langer, C.; Eilers, M.; et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013, 3, e102. [Google Scholar] [CrossRef]
- Milburn, M.V.; Tong, L.; deVos, A.M.; Brunger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef]
- Feig, L.A.; Cooper, G.M. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol. Cell Biol. 1988, 8, 2472–2478. [Google Scholar] [CrossRef]
- Weissbach, S.; Langer, C.; Puppe, B.; Nedeva, T.; Bach, E.; Kull, M.; Bargou, R.; Einsele, H.; Rosenwald, A.; Knop, S.; et al. The molecular spectrum and clinical impact of DIS3 mutations in multiple myeloma. Br. J. Haematol. 2015, 169, 57–70. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef]
- Billadeau, D.; Liu, P.; Jelinek, D.; Shah, N.; LeBien, T.W.; Van Ness, B. Activating mutations in the N- and K-ras oncogenes differentially affect the growth properties of the IL-6-dependent myeloma cell line ANBL6. Cancer Res. 1997, 57, 2268–2275. [Google Scholar] [PubMed]
- Rowley, M.; Liu, P.; Van Ness, B. Heterogeneity in therapeutic response of genetically altered myeloma cell lines to interleukin 6, dexamethasone, doxorubicin, and melphalan. Blood 2000, 96, 3175–3180. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Md Hanafiah, N.H.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Moreau-Aubry, A.; Descamps, G.; Gomez-Bougie, P.; Maiga, S.; Gaignard, A.; Chiron, D.; Menoret, E.; Le Gouill, S.; Moreau, P. Whole-exon sequencing of human myeloma cell lines shows mutations related to myeloma patients at relapse with major hits in the DNA regulation and repair pathways. J. Hematol. Oncol. 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed]
- Hyun, T.; Yam, A.; Pece, S.; Xie, X.; Zhang, J.; Miki, T.; Gutkind, J.S.; Li, W. Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood 2000, 96, 3560–3568. [Google Scholar] [CrossRef]
- Dumaz, N. Mechanism of RAF isoform switching induced by oncogenic RAS in melanoma. Small GTPases 2011, 2, 289–292. [Google Scholar] [CrossRef]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef]
- Keppler, S.; Weissbach, S.; Langer, C.; Knop, S.; Pischimarov, J.; Kull, M.; Stühmer, T.; Steinbrunn, T.; Bargou, R.; Einsele, H.; et al. Rare SNPs in receptor tyrosine kinases are negative outcome predictors in multiple myeloma. Oncotarget 2016, 7, 38762–38774. [Google Scholar] [CrossRef]
- Kropff, M.; Liebisch, P.; Knop, S.; Weisel, K.; Wand, H.; Gann, C.N.; Berdel, W.E.; Einsele, H.; Deutsche Studiengruppe Multiples Myelom. DSMM XI study: Dose definition for intravenous cyclophosphamide in combination with bortezomib/dexamethasone for remission induction in patients with newly diagnosed myeloma. Ann. Hematol. 2009, 88, 1125–1130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stühmer, T.; Arts, J.; Chatterjee, M.; Borawski, J.; Wolff, A.; King, P.; Einsele, H.; Leo, E.; Bargou, R.C. Preclinical anti-myeloma activity of the novel HDAC-inhibitor JNJ-26481585. Br. J. Haematol. 2010, 149, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, C.L.; Leich, E.; Sbiera, S.; Weismann, D.; Rosenwald, A.; Allolio, B.; Fassnacht, M. Single nucleotide polymorphism microarray analysis in cortisol-secreting adrenocortical adenomas identifies new candidate genes and pathways. Neoplasia 2012, 14, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Leich, E.; Liebisch, G.; Sbiera, I.; Schirbel, A.; Wiemer, L.; Matysik, S.; Eckhardt, C.; Gardill, F.; Gehl, A.; et al. Mitotane Inhibits Sterol-O-Acyl Transferase 1 Triggering Lipid-Mediated Endoplasmic Reticulum Stress and Apoptosis in Adrenocortical Carcinoma Cells. Endocrinology 2015, 156, 3895–3908. [Google Scholar] [CrossRef] [PubMed]
- Steinbrunn, T.; Chatterjee, M.; Bargou, R.C.; Stuhmer, T. Efficient transient transfection of human multiple myeloma cells by electroporation—An appraisal. PLoS ONE 2014, 9, e97443. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytogenetic Parameters | KRAS Mut, n = 16 | KRAS WT, n = 64 | p-Value |
|---|---|---|---|
| 13q deletion; no, yes | 9, 7 | 29, 35 | 0.577 |
| 17p deletion; no, yes | 14, 2 | 50, 14 | 0.504 |
| 1q gain; no, yes | 12, 4 | 41, 22 | 0.559 |
| 9q gain; no, yes | 10, 6 | 33, 31 | 0.577 |
| t(4;14); no, yes | 12, 4 | 48, 16 | 1 |
| t(11;14); no, yes | 12, 4 | 49, 15 | 1 |
| t(14;16); no, yes | 15, 1 | 61, 2 | 0.498 |
| t(8;14); no, yes | 14, 2 | 61, 1 | 0.105 |
| t(14;20); no, yes | 16, 0 | 62, 0 | 1 |
| DIS3 mut; no, yes | 15, 1 | 56, 8 | 0.679 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weißbach, S.; Heredia-Guerrero, S.C.; Barnsteiner, S.; Großhans, L.; Bodem, J.; Starz, H.; Langer, C.; Appenzeller, S.; Knop, S.; Steinbrunn, T.; et al. Exon-4 Mutations in KRAS Affect MEK/ERK and PI3K/AKT Signaling in Human Multiple Myeloma Cell Lines. Cancers 2020, 12, 455. https://doi.org/10.3390/cancers12020455
Weißbach S, Heredia-Guerrero SC, Barnsteiner S, Großhans L, Bodem J, Starz H, Langer C, Appenzeller S, Knop S, Steinbrunn T, et al. Exon-4 Mutations in KRAS Affect MEK/ERK and PI3K/AKT Signaling in Human Multiple Myeloma Cell Lines. Cancers. 2020; 12(2):455. https://doi.org/10.3390/cancers12020455
Chicago/Turabian StyleWeißbach, Susann, Sofia Catalina Heredia-Guerrero, Stefanie Barnsteiner, Lukas Großhans, Jochen Bodem, Hanna Starz, Christian Langer, Silke Appenzeller, Stefan Knop, Torsten Steinbrunn, and et al. 2020. "Exon-4 Mutations in KRAS Affect MEK/ERK and PI3K/AKT Signaling in Human Multiple Myeloma Cell Lines" Cancers 12, no. 2: 455. https://doi.org/10.3390/cancers12020455
APA StyleWeißbach, S., Heredia-Guerrero, S. C., Barnsteiner, S., Großhans, L., Bodem, J., Starz, H., Langer, C., Appenzeller, S., Knop, S., Steinbrunn, T., Rost, S., Einsele, H., Bargou, R. C., Rosenwald, A., Stühmer, T., & Leich, E. (2020). Exon-4 Mutations in KRAS Affect MEK/ERK and PI3K/AKT Signaling in Human Multiple Myeloma Cell Lines. Cancers, 12(2), 455. https://doi.org/10.3390/cancers12020455