Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
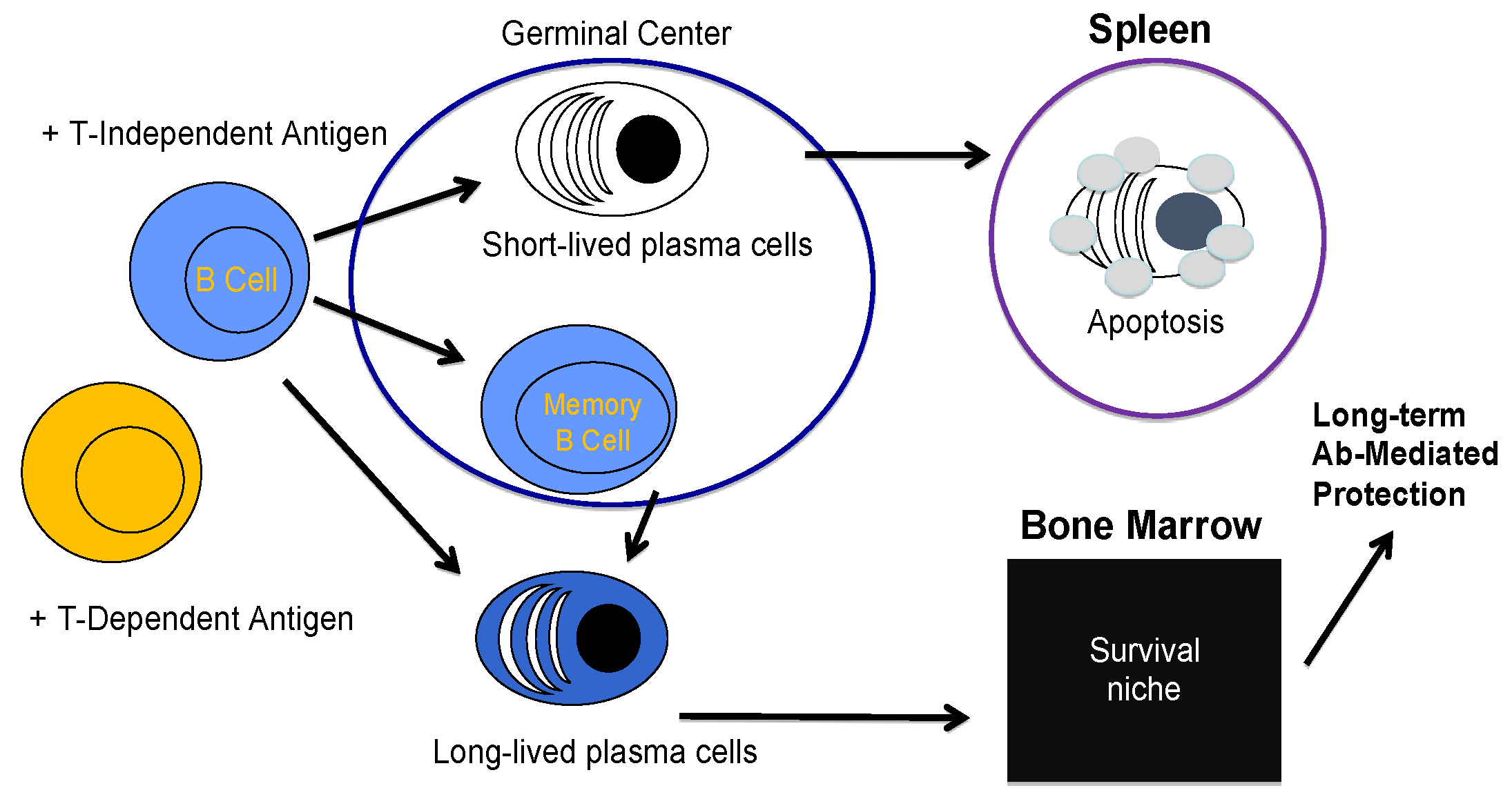
2. B Cell Biology and the Creation of a Plasma Cell
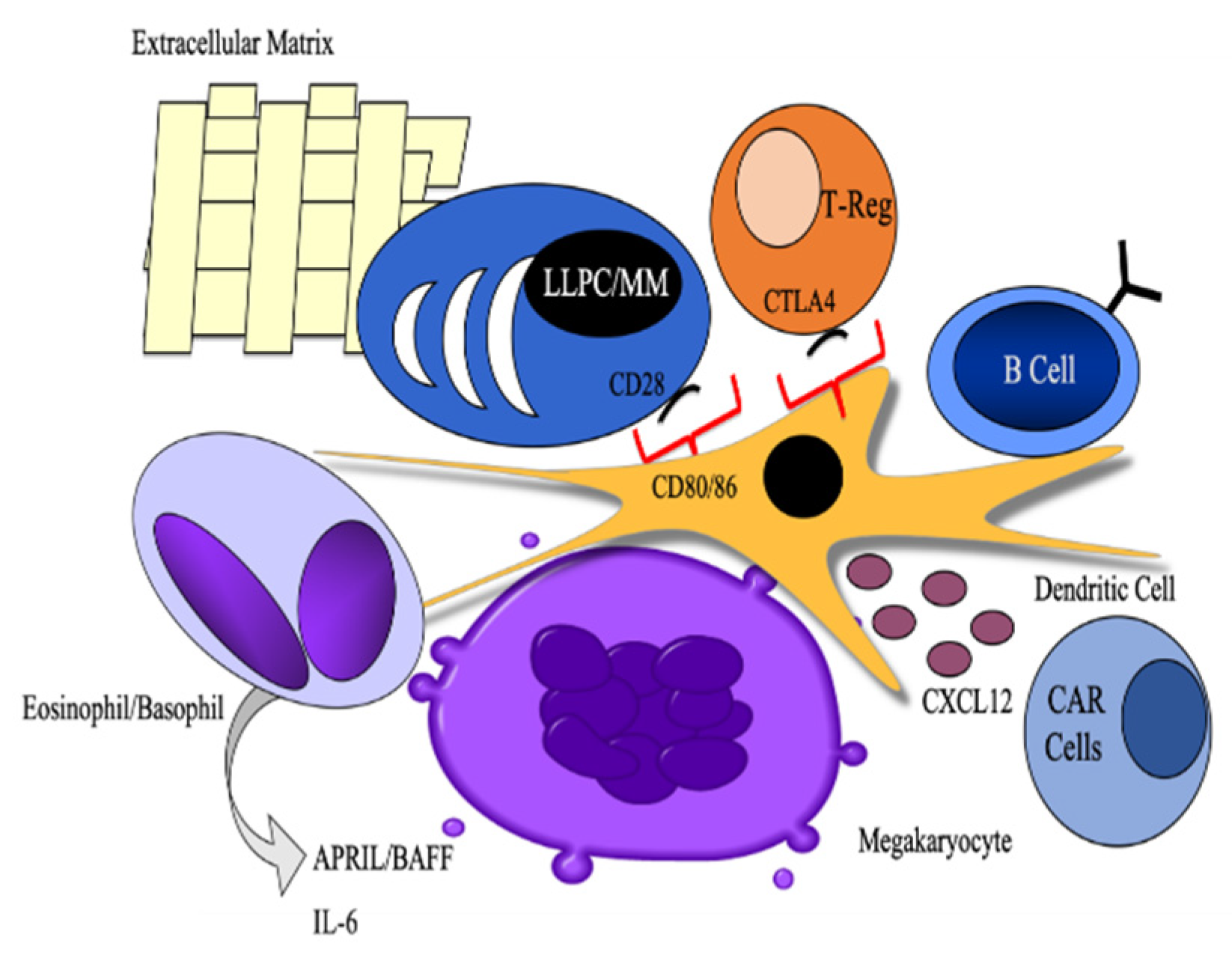
3. Long-Lived Plasma Cell Induction and Maintenance
3.1. Cellular Partners
3.2. Cell Intrinsic Programs
3.2.1. Autophagy
3.2.2. Metabolic Fitness
3.2.3. The Intersection of LLPC and MM
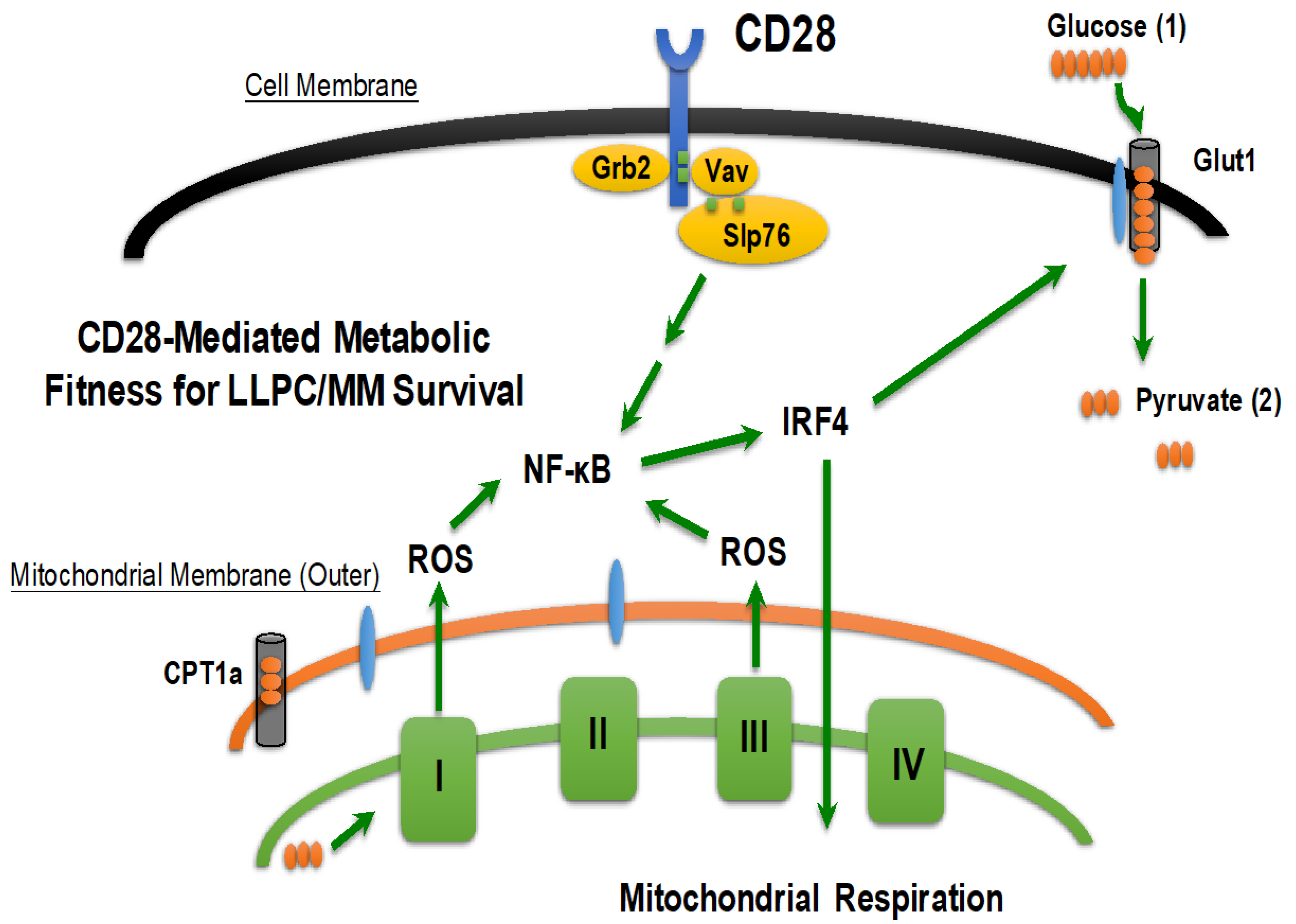
3.2.4. CD28: Bridging the BMME and Intrinsic Survival Programs in LLPC/MM
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kapoor, P.; Rajkumar, S.V. Multiple myeloma in 2016: Fresh perspectives on treatment and moments of clarity. Nat. Rev. Clin. Oncol. 2017, 14, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.Y.; Becker, A.M.; Kennerly, K.M.; Wong, R.; Curtis, J.D.; Llufrio, E.M.; McCommis, K.S.; Fahrmann, J.; Pizzato, H.A.; Nunley, R.M.; et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity 2016, 45, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.Y.; Jash, A.; Yao, C.H.; D’Souza, L.; Wong, R.; Nunley, R.M.; Meares, G.P.; Patti, G.J.; Bhattacharya, D. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep. 2018, 24, 2479–2492.e2476. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- White, R.G. Antibody production by single cells. Nature 1958, 182, 1383–1384. [Google Scholar] [CrossRef]
- Raff, M.C. T and B lymphocytes and immune responses. Nature 1973, 242, 19–23. [Google Scholar] [CrossRef]
- Bjorneboe, M.; Gormsen, H.; Lundquist, F. Further experimental studies on the role of the plasma cells as antibody producers. J. Immunol. 1947, 55, 121–129. [Google Scholar]
- Coutinho, A.; Moller, G. Thymus-independent B-cell induction and paralysis. Adv. Immunol. 1975, 21, 113–236. [Google Scholar]
- Chung, J.B.; Silverman, M.; Monroe, J.G. Transitional B cells: Step by step towards immune competence. Trends Immunol. 2003, 24, 343–349. [Google Scholar] [CrossRef]
- Pelanda, R.; Torres, R.M. Central B-cell tolerance: Where selection begins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007146. [Google Scholar] [CrossRef] [PubMed]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.M.; Martin, F.; Gartland, G.L.; Carter, R.H.; Kearney, J.F. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur. J. Immunol. 1997, 27, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- McHeyzer-Williams, L.J.; Driver, D.J.; McHeyzer-Williams, M.G. Germinal center reaction. Curr. Opin. Hematol. 2001, 8, 52–59. [Google Scholar] [CrossRef]
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242. [Google Scholar] [CrossRef]
- McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005, 23, 487–513. [Google Scholar] [CrossRef]
- Ramadan, A.; Land, W.G.; Paczesny, S. Editorial: Danger Signals Triggering Immune Response and Inflammation. Front. Immunol. 2017, 8, 979. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Lechouane, F.; Bonaud, A.; Delpy, L.; Casola, S.; Oruc, Z.; Chemin, G.; Cogne, M.; Sirac, C. B-cell receptor signal strength influences terminal differentiation. Eur. J. Immunol. 2013, 43, 619–628. [Google Scholar] [CrossRef]
- Chiron, D.; Bekeredjian-Ding, I.; Pellat-Deceunynck, C.; Bataille, R.; Jego, G. Toll-like receptors: Lessons to learn from normal and malignant human B cells. Blood 2008, 112, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Park, E.; Park, H.R.; Jeon, Y.J.; Cha, S.H.; Lee, S.C. Antioxidant activity of far-infrared radiated rice hull extracts on reactive oxygen species scavenging and oxidative DNA damage in human lymphocytes. J. Med. Food 2006, 9, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.A.; Crapo, J.D. Biology of disease: Free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Price, M.J.; Patterson, D.G.; Scharer, C.D.; Boss, J.M. Progressive Upregulation of Oxidative Metabolism Facilitates Plasmablast Differentiation to a T-Independent Antigen. Cell Rep. 2018, 23, 3152–3159. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Yin, Q.; Fujio, Y.; Walsh, K.; Griendling, K.K. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 22699–22704. [Google Scholar] [CrossRef]
- Marumo, T.; Schini-Kerth, V.B.; Fisslthaler, B.; Busse, R. Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-kappaB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation 1997, 96, 2361–2367. [Google Scholar] [CrossRef]
- Zhu, Z.; Shukla, A.; Ramezani-Rad, P.; Apgar, J.R.; Rickert, R.C. The AKT isoforms 1 and 2 drive B cell fate decisions during the germinal center response. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef]
- Higgins, B.W.; McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Programming Isotype-Specific Plasma Cell Function. Trends Immunol. 2019, 40, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.; Winslow, G.M. IgM in microbial infections: Taken for granted? Immunol. Lett. 2009, 125, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, C.M.; Falcone, F.H.; Dunne, D.W. Helminth Allergens, Parasite-Specific IgE, and Its Protective Role in Human Immunity. Front. Immunol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef]
- Nakajima, A.; Vogelzang, A.; Maruya, M.; Miyajima, M.; Murata, M.; Son, A.; Kuwahara, T.; Tsuruyama, T.; Yamada, S.; Matsuura, M.; et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med. 2018, 215, 2019–2034. [Google Scholar] [CrossRef]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002, 298, 2199–2202. [Google Scholar] [CrossRef]
- Ahuja, A.; Anderson, S.M.; Khalil, A.; Shlomchik, M.J. Maintenance of the plasma cell pool is independent of memory B cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4802–4807. [Google Scholar] [CrossRef]
- Gray, D.; Skarvall, H. B-cell memory is short-lived in the absence of antigen. Nature 1988, 336, 70–73. [Google Scholar] [CrossRef]
- Manz, R.A.; Lohning, M.; Cassese, G.; Thiel, A.; Radbruch, A. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 1998, 10, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.L.; Sell, S. The Immunoglobulins of Mice. V. The Metabolic (Catabolic) Properties of Five Immunoglobulin Classes. J. Exp. Med. 1965, 122, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dorner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Tellier, J.; Nutt, S.L. Standing out from the crowd: How to identify plasma cells. Eur. J. Immunol. 2017, 47, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.L.; Peel, J.N.; Scharer, C.D.; Risley, C.A.; Chisolm, D.A.; Schultz, M.D.; Yu, B.; Ballesteros-Tato, A.; Wojciechowski, W.; Mousseau, B.; et al. T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-gamma on B Cells. Immunity 2019, 50, 1172–1187 e1177. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Emslie, D.; Shi, W.; Kratina, T.; Wellard, C.; Karnowski, A.; Erikci, E.; Smyth, G.K.; Chowdhury, K.; Tarlinton, D.; et al. The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. J. Exp. Med. 2014, 211, 827–840. [Google Scholar] [CrossRef]
- Chevrier, S.; Genton, C.; Kallies, A.; Karnowski, A.; Otten, L.A.; Malissen, B.; Malissen, M.; Botto, M.; Corcoran, L.M.; Nutt, S.L.; et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc. Natl. Acad. Sci. USA 2009, 106, 3895–3900. [Google Scholar] [CrossRef]
- Barwick, B.G.; Scharer, C.D.; Bally, A.P.R.; Boss, J.M. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat. Immunol. 2016, 17, 1216–1225. [Google Scholar] [CrossRef]
- Scharer, C.D.; Barwick, B.G.; Guo, M.; Bally, A.P.R.; Boss, J.M. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat. Commun. 2018, 9, 1698. [Google Scholar] [CrossRef]
- Delogu, A.; Schebesta, A.; Sun, Q.; Aschenbrenner, K.; Perlot, T.; Busslinger, M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity 2006, 24, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Bohannon, C.; Powers, R.; Satyabhama, L.; Cui, A.; Tipton, C.; Michaeli, M.; Skountzou, I.; Mittler, R.S.; Kleinstein, S.H.; Mehr, R.; et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat. Commun. 2016, 7, 11826. [Google Scholar] [CrossRef] [PubMed]
- Landsverk, O.J.; Snir, O.; Casado, R.B.; Richter, L.; Mold, J.E.; Reu, P.; Horneland, R.; Paulsen, V.; Yaqub, S.; Aandahl, E.M.; et al. Antibody-secreting plasma cells persist for decades in human intestine. J. Exp. Med. 2017, 214, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Luger, E.O.; Fokuhl, V.; Wegmann, M.; Abram, M.; Tillack, K.; Achatz, G.; Manz, R.A.; Worm, M.; Radbruch, A.; Renz, H. Induction of long-lived allergen-specific plasma cells by mucosal allergen challenge. J. Allergy Clin. Immunol. 2009, 124, 819–826 e814. [Google Scholar] [CrossRef]
- Nie, Y.; Waite, J.; Brewer, F.; Sunshine, M.J.; Littman, D.R.; Zou, Y.R. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J. Exp. Med. 2004, 200, 1145–1156. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Hyman, P.L.; Lu, T.T.; Ngo, V.N.; Bidgol, A.; Suzuki, G.; Zou, Y.R.; Littman, D.R.; Cyster, J.G. A coordinated change in chemokine responsiveness guides plasma cell movements. J. Exp. Med. 2001, 194, 45–56. [Google Scholar] [CrossRef]
- Belnoue, E.; Tougne, C.; Rochat, A.F.; Lambert, P.H.; Pinschewer, D.D.; Siegrist, C.A. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J. Immunol. 2012, 188, 1283–1291. [Google Scholar] [CrossRef]
- Chu, V.T.; Frohlich, A.; Steinhauser, G.; Scheel, T.; Roch, T.; Fillatreau, S.; Lee, J.J.; Lohning, M.; Berek, C. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat. Immunol. 2011, 12, 151–159. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Garimalla, S.; Xiao, H.; Kyu, S.; Albizua, I.; Galipeau, J.; Chiang, K.Y.; Waller, E.K.; Wu, R.; Gibson, G.; et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat. Commun. 2018, 9, 3698. [Google Scholar] [CrossRef]
- Minges Wols, H.A.; Underhill, G.H.; Kansas, G.S.; Witte, P.L. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J. Immunol. 2002, 169, 4213–4221. [Google Scholar] [CrossRef]
- Jourdan, M.; Cren, M.; Robert, N.; Bollore, K.; Fest, T.; Duperray, C.; Guilloton, F.; Hose, D.; Tarte, K.; Klein, B. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia 2014, 28, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Glatman Zaretsky, A.; Konradt, C.; Depis, F.; Wing, J.B.; Goenka, R.; Atria, D.G.; Silver, J.S.; Cho, S.; Wolf, A.I.; Quinn, W.J.; et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep. 2017, 18, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Koorella, C.; Nair, J.R.; Murray, M.E.; Carlson, L.M.; Watkins, S.K.; Lee, K.P. Novel regulation of CD80/CD86-induced phosphatidylinositol 3-kinase signaling by NOTCH1 protein in interleukin-6 and indoleamine 2,3-dioxygenase production by dendritic cells. J. Biol. Chem. 2014, 289, 7747–7762. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Gomez, M.; Talke, Y.; Goebel, N.; Hermann, F.; Reich, B.; Mack, M. Basophils support the survival of plasma cells in mice. J. Immunol. 2010, 185, 7180–7185. [Google Scholar] [CrossRef]
- Winter, O.; Moser, K.; Mohr, E.; Zotos, D.; Kaminski, H.; Szyska, M.; Roth, K.; Wong, D.M.; Dame, C.; Tarlinton, D.M.; et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 2010, 116, 1867–1875. [Google Scholar] [CrossRef]
- Minges Wols, H.A.; Ippolito, J.A.; Yu, Z.; Palmer, J.L.; White, F.A.; Le, P.T.; Witte, P.L. The effects of microenvironment and internal programming on plasma cell survival. Int. Immunol. 2007, 19, 837–846. [Google Scholar] [CrossRef]
- Mohr, E.; Serre, K.; Manz, R.A.; Cunningham, A.F.; Khan, M.; Hardie, D.L.; Bird, R.; MacLennan, I.C. Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J. Immunol. 2009, 182, 2113–2123. [Google Scholar] [CrossRef]
- Benson, M.J.; Dillon, S.R.; Castigli, E.; Geha, R.S.; Xu, S.; Lam, K.P.; Noelle, R.J. Cutting edge: The dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J. Immunol. 2008, 180, 3655–3659. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.H.; Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003, 194, 29–38. [Google Scholar] [CrossRef]
- Gass, J.N.; Gunn, K.E.; Sriburi, R.; Brewer, J.W. Stressed-out B cells? Plasma-cell differentiation and the unfolded protein response. Trends Immunol. 2004, 25, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Stromberg, T.; Dimberg, A.; Hammarberg, A.; Carlson, K.; Osterborg, A.; Nilsson, K.; Jernberg-Wiklund, H. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood 2004, 103, 3138–3147. [Google Scholar] [CrossRef]
- Seo, W.D.; Lee, J.H.; Jia, Y.; Wu, C.; Lee, S.J. Saponarin activates AMPK in a calcium-dependent manner and suppresses gluconeogenesis and increases glucose uptake via phosphorylation of CRTC2 and HDAC5. Bioorg. Med. Chem. Lett. 2015, 25, 5237–5242. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, J.; Mao, K.; She, H.; Ren, Y.; Gui, C.; Wu, X.; Zou, F.; Li, W. Mitochondrial calcium dysfunction contributes to autophagic cell death induced by MPP(+) via AMPK pathway. Biochem. Biophys. Res. Commun. 2019, 509, 390–394. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, J.E.; Samelson, L.E. T cell antigen-receptor signal transduction. Curr. Opin. Immunol. 1999, 11, 242–248. [Google Scholar] [CrossRef]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes—New targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1beta regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595. [Google Scholar] [CrossRef]
- Boise, L.H.; Shanmugam, M. Stromal Support of Metabolic Function through Mitochondrial Transfer in Multiple Myeloma. Cancer Res. 2019, 79, 2102–2103. [Google Scholar] [CrossRef]
- Richardson, P.G. A review of the proteasome inhibitor bortezomib in multiple myeloma. Expert. Opin. Pharmacother. 2004, 5, 1321–1331. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Gado, K.; Domjan, G.; Hegyesi, H.; Falus, A. Role of INTERLEUKIN-6 in the pathogenesis of multiple myeloma. Cell Biol. Int. 2000, 24, 195–209. [Google Scholar] [CrossRef]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Langat, D.L.; Wheaton, D.A.; Platt, J.S.; Sifers, T.; Hunt, J.S. Signaling pathways for B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) in human placenta. Am. J. Pathol. 2008, 172, 1303–1311. [Google Scholar] [CrossRef]
- Bossen, C.; Schneider, P. BAFF, APRIL and their receptors: Structure, function and signaling. Semin. Immunol. 2006, 18, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682. [Google Scholar] [CrossRef]
- Tai, Y.T.; Podar, K.; Catley, L.; Tseng, Y.H.; Akiyama, M.; Shringarpure, R.; Burger, R.; Hideshima, T.; Chauhan, D.; Mitsiades, N.; et al. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3’-kinase/AKT signaling. Cancer Res. 2003, 63, 5850–5858. [Google Scholar]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef]
- Gupta, V.A.; Matulis, S.M.; Conage-Pough, J.E.; Nooka, A.K.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017, 129, 1969–1979. [Google Scholar] [CrossRef]
- Peperzak, V.; Vikstrom, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget 2019, 10, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; Lammerts van Bueren, J.J.; Parren, P.W.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Gogishvili, T.; Danhof, S.; Prommersberger, S.; Rydzek, J.; Schreder, M.; Brede, C.; Einsele, H.; Hudecek, M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7(+) normal lymphocytes. Blood 2017, 130, 2838–2847. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front. Immunol. 2018, 9, 2551. [Google Scholar] [CrossRef]
- Abdallah, N.; Kumar, S.K. Daratumumab in untreated newly diagnosed multiple myeloma. Ther. Adv. Hematol. 2019, 10, 2040620719894871. [Google Scholar] [CrossRef]
- Underhill, G.H.; George, D.; Bremer, E.G.; Kansas, G.S. Gene expression profiling reveals a highly specialized genetic program of plasma cells. Blood 2003, 101, 4013–4021. [Google Scholar] [CrossRef]
- Tarte, K.; Zhan, F.; De Vos, J.; Klein, B.; Shaughnessy, J., Jr. Gene expression profiling of plasma cells and plasmablasts: Toward a better understanding of the late stages of B-cell differentiation. Blood 2003, 102, 592–600. [Google Scholar] [CrossRef]
- Tellier, J.; Shi, W.; Minnich, M.; Liao, Y.; Crawford, S.; Smyth, G.K.; Kallies, A.; Busslinger, M.; Nutt, S.L. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 2016, 17, 323–330. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- June, C.H.; Ledbetter, J.A.; Gillespie, M.M.; Lindsten, T.; Thompson, C.B. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell Biol. 1987, 7, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, E.A.; Nguyen, K.A.; Kuchroo, V.K. CD28/B7 costimulation: A review. Crit. Rev. Immunol. 1998, 18, 389–418. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.L.; Frauwirth, K.A.; Thompson, C.B. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001, 1, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Brady, W.; Grosmaire, L.; Aruffo, A.; Damle, N.K.; Ledbetter, J.A. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J. Exp. Med. 1991, 173, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Vella, A.T.; Mitchell, T.; Groth, B.; Linsley, P.S.; Green, J.M.; Thompson, C.B.; Kappler, J.W.; Marrack, P. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J. Immunol. 1997, 158, 4714–4720. [Google Scholar]
- Rudd, C.E. Upstream-downstream: CD28 cosignaling pathways and T cell function. Immunity 1996, 4, 527–534. [Google Scholar] [CrossRef]
- Pellat-Deceunynck, C.; Bataille, R.; Robillard, N.; Harousseau, J.L.; Rapp, M.J.; Juge-Morineau, N.; Wijdenes, J.; Amiot, M. Expression of CD28 and CD40 in human myeloma cells: A comparative study with normal plasma cells. Blood 1994, 84, 2597–2603. [Google Scholar] [CrossRef]
- Zhang, X.G.; Olive, D.; Devos, J.; Rebouissou, C.; Ghiotto-Ragueneau, M.; Ferlin, M.; Klein, B. Malignant plasma cell lines express a functional CD28 molecule. Leukemia 1998, 12, 610–618. [Google Scholar] [CrossRef]
- Kozbor, D.; Moretta, A.; Messner, H.A.; Moretta, L.; Croce, C.M. Tp44 molecules involved in antigen-independent T cell activation are expressed on human plasma cells. J. Immunol. 1987, 138, 4128–4132. [Google Scholar]
- Bahlis, N.J.; King, A.M.; Kolonias, D.; Carlson, L.M.; Liu, H.Y.; Hussein, M.A.; Terebelo, H.R.; Byrne, G.E., Jr.; Levine, B.L.; Boise, L.H.; et al. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood 2007, 109, 5002–5010. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Gavile, C.M.; Nair, J.R.; Koorella, C.; Carlson, L.M.; Buac, D.; Utley, A.; Chesi, M.; Bergsagel, P.L.; Boise, L.H.; et al. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 2014, 123, 3770–3779. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Dodson, L.F.; Boomer, J.S.; Deppong, C.M.; Shah, D.D.; Sim, J.; Bricker, T.L.; Russell, J.H.; Green, J.M. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol. Cell Biol. 2009, 29, 3710–3721. [Google Scholar] [CrossRef]
- Ferguson, S.E.; Han, S.; Kelsoe, G.; Thompson, C.B. CD28 is required for germinal center formation. J. Immunol. 1996, 156, 4576–4581. [Google Scholar]
- Friend, L.D.; Shah, D.D.; Deppong, C.; Lin, J.; Bricker, T.L.; Juehne, T.I.; Rose, C.M.; Green, J.M. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J. Exp. Med. 2006, 203, 2121–2133. [Google Scholar] [CrossRef]
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kundig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612. [Google Scholar] [CrossRef]
- Van Wijk, F.; Nierkens, S.; de Jong, W.; Wehrens, E.J.; Boon, L.; Van Kooten, P.; Knippels, L.M.; Pieters, R. The CD28/CTLA-4-B7 signaling pathway is involved in both allergic sensitization and tolerance induction to orally administered peanut proteins. J. Immunol. 2007, 178, 6894–6900. [Google Scholar] [CrossRef]
- Ribeiro, A.C.; Laurindo, I.M.; Guedes, L.K.; Saad, C.G.; Moraes, J.C.; Silva, C.A.; Bonfa, E. Abatacept severely reduces the immune response to pandemic 2009 influenza A/H1N1 vaccination in patients with rheumatoid arthritis. Arthritis Care Res. (Hoboken.) 2012. [Google Scholar] [CrossRef]
- Phelps, C.J.; Ball, S.F.; Vaught, T.D.; Vance, A.M.; Mendicino, M.; Monahan, J.A.; Walters, A.H.; Wells, K.D.; Dandro, A.S.; Ramsoondar, J.J.; et al. Production and characterization of transgenic pigs expressing porcine CTLA4-Ig. Xenotransplantation 2009, 16, 477–485. [Google Scholar] [CrossRef]
- Horspool, J.H.; Perrin, P.J.; Woodcock, J.B.; Cox, J.H.; King, C.L.; June, C.H.; Harlan, D.M.; St Louis, D.C.; Lee, K.P. Nucleic acid vaccine-induced immune responses require CD28 costimulation and are regulated by CTLA4. J. Immunol. 1998, 160, 2706–2714. [Google Scholar] [PubMed]
- Borriello, F.; Sethna, M.P.; Boyd, S.D.; Schweitzer, A.N.; Tivol, E.A.; Jacoby, D.; Strom, T.B.; Simpson, E.M.; Freeman, G.J.; Sharpe, A.H. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity 1997, 6, 303–313. [Google Scholar] [CrossRef]
- Akalin, E.; Chandraker, A.; Russell, M.E.; Turka, L.A.; Hancock, W.W.; Sayegh, M.H. CD28-B7 T cell costimulatory blockade by CTLA4Ig in the rat renal allograft model: Inhibition of cell-mediated and humoral immune responses in vivo. Transplantation 1996, 62, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Rathmell, J.C. The metabolic life and times of a T-cell. Immunol. Rev. 2010, 236, 190–202. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Los, M.; Schenk, H.; Hexel, K.; Baeuerle, P.A.; Droge, W.; Schulze-Osthoff, K. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995, 14, 3731–3740. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.J.; Gerondakis, S. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: Modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J. Exp. Med. 2000, 191, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, J.; Schumacher, V.; Ahrens, S.; Kading, N.; Feldhoff, L.M.; Huber, M.; Rupp, J.; Raczkowski, F.; Mittrucker, H.W. Interferon Regulatory Factor 4 controls TH1 cell effector function and metabolism. Sci. Rep. 2016, 6, 35521. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Miasari, M.; Shi, W.; Xin, A.; Henstridge, D.C.; Preston, S.; Pellegrini, M.; Belz, G.T.; Smyth, G.K.; Febbraio, M.A.; et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 2013, 14, 1155–1165. [Google Scholar] [CrossRef]
- Rozanski, C.H.; Arens, R.; Carlson, L.M.; Nair, J.; Boise, L.H.; Chanan-Khan, A.A.; Schoenberger, S.P.; Lee, K.P. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J. Exp. Med. 2011, 208, 1435–1446. [Google Scholar] [CrossRef]
- Boomer, J.S.; Green, J.M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef]
- Utley, A.; Chavel, C.; Lightman, S.; Holling, G.A.; Cooper, J.; Peng, P.; Liu, W.; Barwick, B.G.; Gavile, C.M.; Maguire, O.; et al. CD28 Regulates Metabolic Fitness for Long-Lived Plasma Cell Survival. Cell Rep. 2020, 31, 107815. [Google Scholar] [CrossRef]
- Gavile, C.M.; Barwick, B.G.; Newman, S.; Neri, P.; Nooka, A.K.; Lonial, S.; Lee, K.P.; Boise, L.H. CD86 regulates myeloma cell survival. Blood Adv. 2017, 1, 2307–2319. [Google Scholar] [CrossRef]
- Boise, L.H.; Kaufman, J.L.; Bahlis, N.J.; Lonial, S.; Lee, K.P. The Tao of myeloma. Blood 2014, 124, 1873–1879. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utley, A.; Lipchick, B.; Lee, K.P.; Nikiforov, M.A. Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells. Cancers 2020, 12, 2117. https://doi.org/10.3390/cancers12082117
Utley A, Lipchick B, Lee KP, Nikiforov MA. Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells. Cancers. 2020; 12(8):2117. https://doi.org/10.3390/cancers12082117
Chicago/Turabian StyleUtley, Adam, Brittany Lipchick, Kelvin P. Lee, and Mikhail A. Nikiforov. 2020. "Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells" Cancers 12, no. 8: 2117. https://doi.org/10.3390/cancers12082117
APA StyleUtley, A., Lipchick, B., Lee, K. P., & Nikiforov, M. A. (2020). Targeting Multiple Myeloma through the Biology of Long-Lived Plasma Cells. Cancers, 12(8), 2117. https://doi.org/10.3390/cancers12082117