Multiple Myeloma: Available Therapies and Causes of Drug Resistance


 ,
,  , ,
, ,
Abstract
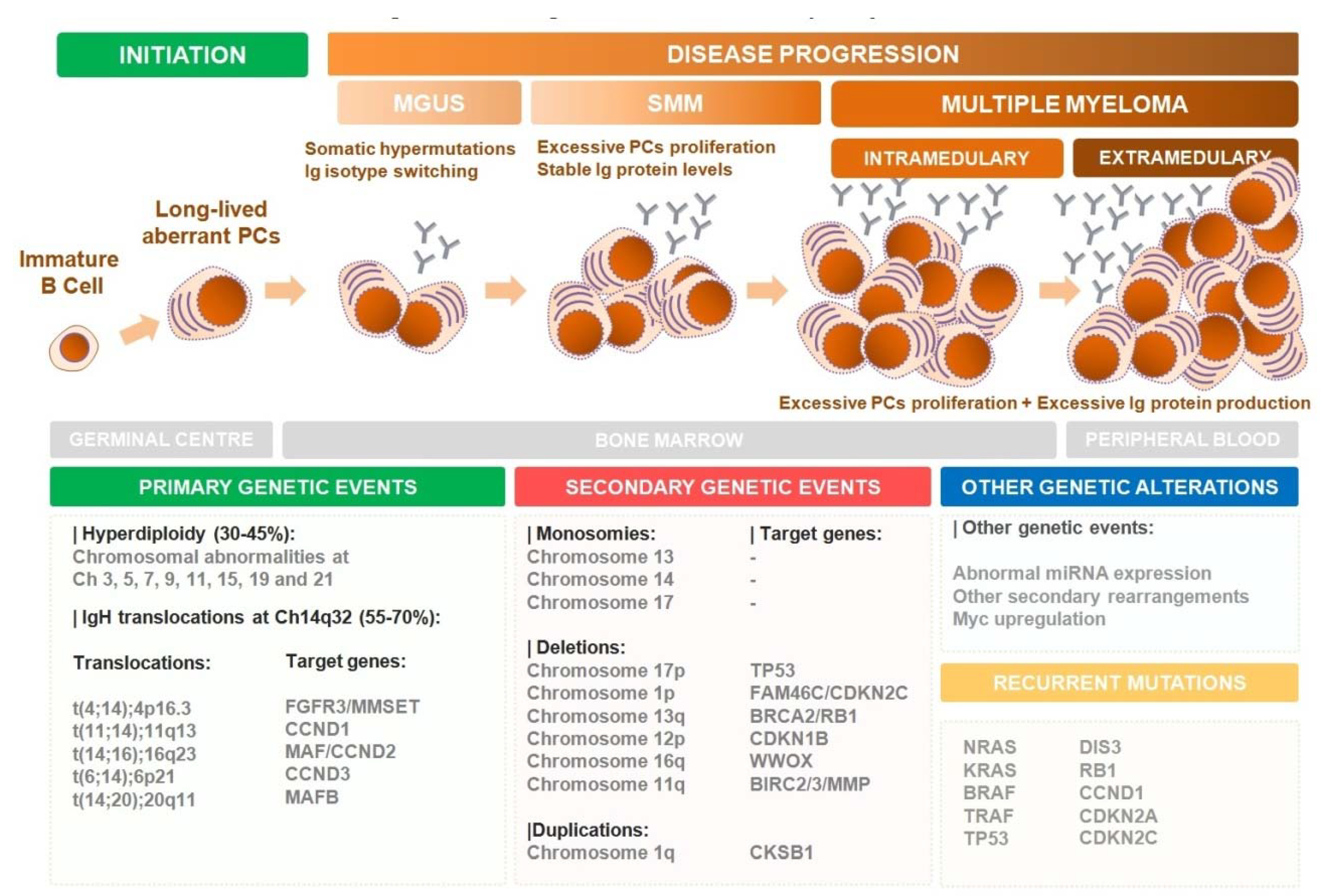
1. Introduction
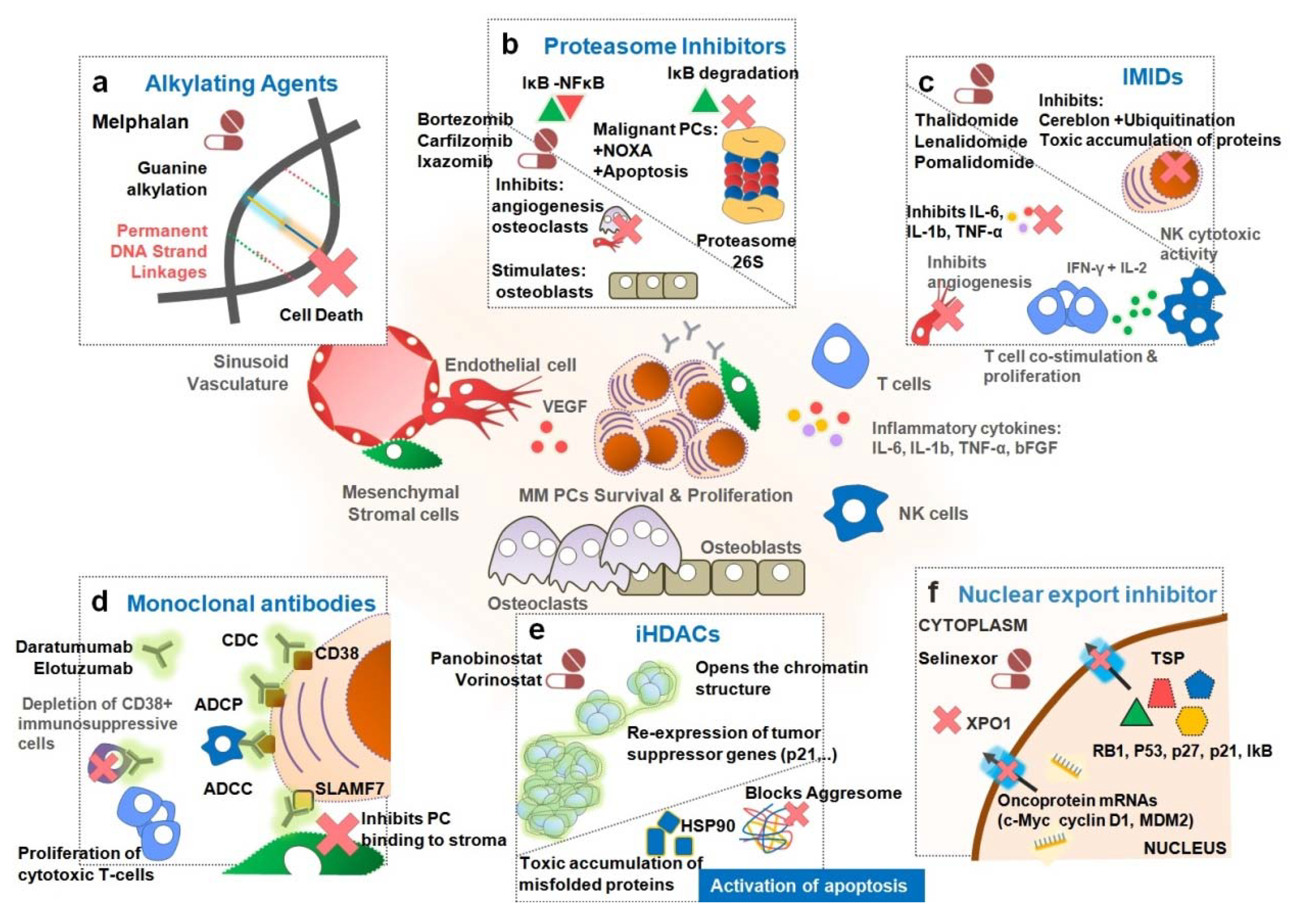
2. Treatment of Multiple Myeloma
2.1. Proteasome Inhibitors (PIs)
2.1.1. Bortezomib
2.1.2. Carfilzomib
2.1.3. Ixazomib
2.2. Immunomodulatory Drugs (IMIDs)
2.2.1. Thalidomide
2.2.2. Lenalidomide
2.2.3. Pomalidomide
2.3. Monoclonal Antibodies (mAbs)
2.3.1. Anti-CD38
2.3.2. Elotuzumab
2.4. Histone Deacetylase Inhibitors (iHDACs)
2.4.1. Panobinostat
2.4.2. Vorinostat
2.5. Other Drugs
Selinexor
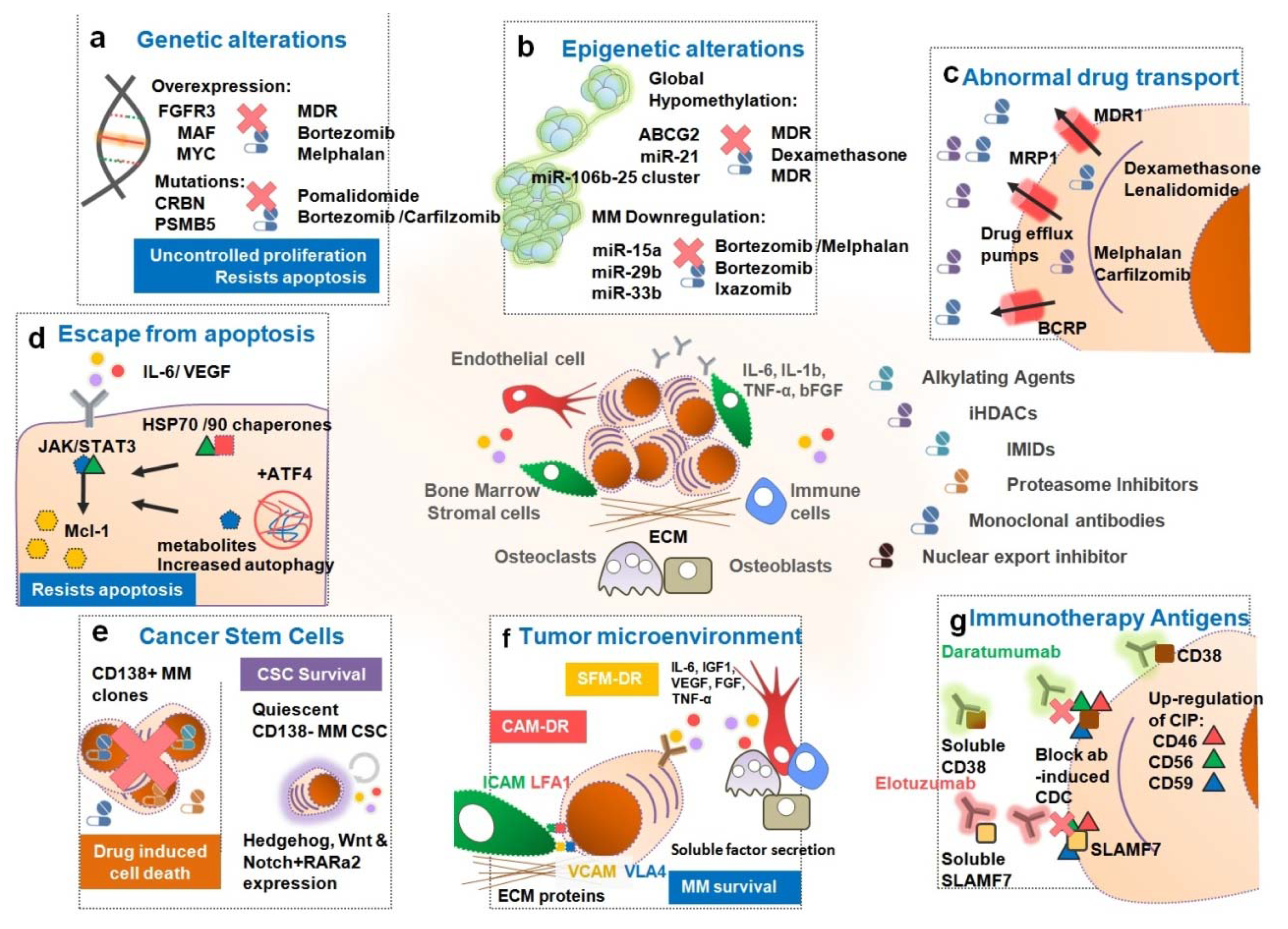
3. Causes of Drug Resistance in MM Patients
3.1. Genetic Alterations Influencing Drug Resistance in MM
3.2. Epigenetic Alterations and MicroRNAs Responsible for Drug Resistance in MM
3.3. Abnormal Drug Transport
3.4. Escape from Apoptosis, Autophagy Activation and Dysregulated Intracellular Signaling Pathways
3.5. Persistence of Cancer Stem Cells
3.6. Tumor Microenvironment
3.7. Other Specific Mechanisms for Immunotherapies with Antibodies
4. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers. 2017, 3, nrdp201746. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, V.S.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 Patients with Newly Diagnosed Multiple Myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Graubard, B.; Katzmann, J.; Kyle, R.; Ahmadizadeh, I.; Clark, R.; Kumar, S.; Dispenzieri, A.; Greenberg, A.; Therneau, T.; et al. Racial disparities in the prevalence of monoclonal gammopathies: A population-based study of 12 482 persons from the National Health and Nutritional Examination Survey. Leukemia 2014, 28, 1537–1542. [Google Scholar] [CrossRef]
- Kumar, S.; Dispenzieri, A.; Lacy, M.; Gertz, M.; Buadi, F.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef]
- Costa, L.J.; Brill, I.K.; Omel, J.; Godby, K.; Kumar, S.K.; Brown, E.E. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv. 2017, 1, 282–287. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.S. The multiple myelomas—current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef]
- Rajkumar, V.S. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 1091–1110. [Google Scholar] [CrossRef]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of Myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef]
- Hauser, A.; Muehlinghaus, G.; Manz, R.; Cassese, G.; Arce, S.; Debes, G.; Hamann, A.; Berek, C.; Lindenau, S.; Doeer, T.; et al. Long-Lived Plasma Cells in Immunity and Inflammation. Ann. N. Y. Acad. Sci. 2003, 987, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [PubMed]
- du Pont, S.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of Multiple Myeloma. J. Clin. Oncol. 2017, 35, JCO.2016.70.670. [Google Scholar]
- Prideaux, S.M.; O’Brien, E.; Chevassut, T.J. The Genetic Architecture of Multiple Myeloma. Adv. Hematol. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.-T.; Mudie, L.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Alzrigat, M.; Párraga, A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, M.W. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, V.S. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. New Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S.; Landgren, O.; Mateos, M.-V. Smoldering multiple myeloma. Blood 2015, 125, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, T.; Kryukov, F.; Rihova, L.; Hajek, R. Plasma cell leukemia: from biology to treatment. Eur. J. Haematol. 2015, 95, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.; Kumar, S.K.; Roeker, L.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Go, R.S.; Dispenzieri, A.; Kapoor, P.; Lust, J.A.; et al. Revised diagnostic criteria for plasma cell leukemia: results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018, 8, 116. [Google Scholar] [CrossRef]
- Cejalvo, M.J.; de la Rubia, J. Which therapies will move to the front line for multiple myeloma? Expert Rev. Hematol. 2017, 10, 383–392. [Google Scholar] [CrossRef]
- Rajkumar, V.S. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37, 62–65. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, V.S. Treatment of Multiple Myeloma: A Comprehensive Review. Clin. Lymphoma Myeloma 2009, 9, 278–288. [Google Scholar] [CrossRef]
- Harousseau, J.-L.; Moreau, P. Autologous Hematopoietic Stem-Cell Transplantation for Multiple Myeloma. New Engl. J. Med. 2009, 360, 2645–2654. [Google Scholar] [CrossRef]
- Mina, R.; Bringhen, S.; Wildes, T.M.; Zweegman, S.; Rosko, A.E. Approach to the Older Adult With Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2019, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S. Treatment of multiple myeloma. Nat. Rev. Clin. Oncol. 2011, 8, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lee, J.; Lahuerta, J.; Morgan, G.; Richardson, P.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012, 26, 149. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.; Kumar, S.; Orlowski, R.; Cook, G.; Richardson, P.; Gertz, M.; Giralt, S.; Mateos, M.; Leleu, X.; Anderson, K. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 2017, 32, 252. [Google Scholar] [CrossRef]
- Giuliani, N.; Accardi, F.; Marchica, V.; Palma, B.; Storti, P.; Toscani, D.; Vicario, E.; Malavasi, F. Novel targets for the treatment of relapsing multiple myeloma. Expert Rev. Hematol. 2019, 12, 481–496. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Facon, T. Frontline therapy of multiple myeloma. Blood 2015, 125, 3076–3084. [Google Scholar] [CrossRef]
- Kumar, S.K.; Buadi, F.K.; Rajkumar, V.S. Pros and cons of frontline autologous transplant in multiple myeloma: the debate over timing. Blood 2019, 133, 652–659. [Google Scholar] [CrossRef]
- Palumbo, A.; Bringhen, S.; Ludwig, H.; Dimopoulos, M.A.; Bladé, J.; Mateos, M.V.; Rosiñol, L.; Boccadoro, M.; Cavo, M.; Lokhorst, H.; et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood 2011, 118, 4519–4529. [Google Scholar] [CrossRef]
- Marini, C.; Maia, T.; Bergantim, R.; Pires, J.; Aguiar, E.; Guimarães, J.; Trigo, F. Real-life data on safety and efficacy of autologous stem cell transplantation in elderly patients with multiple myeloma. Ann. Hematol. 2019, 98, 369–379. [Google Scholar] [CrossRef]
- Chng, W.; Dispenzieri, A.; Chim, C.-S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Boise, L.H.; Kaufman, J. How I treat high-risk myeloma. Blood 2015, 126, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Lonial, S. New Targets and New Agents in High-Risk Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2016, e431–e441. [Google Scholar] [CrossRef] [PubMed]
- Mikhael, J.; Ismaila, N.; Cheung, M.C.; Costello, C.; Dhodapkar, M.V.; Kumar, S.; Lacy, M.; Lipe, B.; Little, R.F.; Nikonova, A.; et al. Treatment of Multiple Myeloma: ASCO and CCO Joint Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, JCO.18.02096. [Google Scholar] [CrossRef]
- Moreau, P.; Miguel, S.J.; Sonneveld, P.; Mateos, M.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.; Ludwig, H.; Einsele, H.; et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv52–iv61. [Google Scholar] [CrossRef]
- Harousseau, J. How to select among available options for the treatment of multiple myeloma. Ann. Oncol. 2012, 23, x334–x338. [Google Scholar] [CrossRef]
- Bladé, J.; Rosiñol, L.; de Larrea, C. How I treat relapsed myeloma. Blood 2015, 125, 1532–1540. [Google Scholar] [CrossRef][Green Version]
- Sonneveld, P.; Broijl, A. Treatment of relapsed and refractory multiple myeloma. Haematologica 2016, 101, 396–406. [Google Scholar] [CrossRef]
- Cavo, M. Facing lenalidomide-refractory myeloma. Blood 2019, 134, 99–101. [Google Scholar] [CrossRef]
- Kumar, S.K. Recycling therapies for myeloma: The need for prospective trials. Cancer 2019, 125, 2920–2922. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. New Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Tar. 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, V.S.; Alovic, G.; Alsina, M.; Alexanian, R.; et al. A Phase 2 Study of Bortezomib in Relapsed, Refractory Myeloma. New Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Mohty, M.; Malard, F.; Mohty, B.; Savani, B.; Moreau, P.; Terpos, E. The effects of bortezomib on bone disease in patients with multiple myeloma. Cancer 2014, 120, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Wang, M.; Kaufman, J.L.; Lonial, S.; Jakubowiak, A.J.; Stewart, K.A.; Kukreti, V.; Jagannath, S.; Nagh, K.T.; Alsina, M.; et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012, 119, 5661–5670. [Google Scholar] [CrossRef]
- Stewart, K.A.; Rajkumar, V.S.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. New Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Dytfeld, D.; Griffith, K.A.; Lebovic, D.; Vesole, D.H.; Jagannath, S.; Al-Zoubi, A.; Anderson, T.; Nordgren, B.; Detweiler-Short, K.; et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012, 120, 1801–1809. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors – molecular basis and current perspectives in multiple myeloma. J. Cell Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. New Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Berdeja, J.G.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, K.A.; Hari, P.; Roy, V.; Vescio, R.; et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: An open-label phase 1/2 study. Lancet Oncol. 2014, 15, 1503–1512. [Google Scholar] [CrossRef]
- Kotla, V.; Goel, S.; Nischal, S.; Heuck, C.; Vivek, K.; Das, B.; Verma, A. Mechanism of action of lenalidomide in hematological malignancies. J. Hematol. Oncol. 2009, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C. Lenalidomide and Thalidomide: Mechanisms of Action—Similarities and Differences. Semin. Hematol. 2005, 42, S3–S8. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Shima, Y.; Raje, N.; Davies, F.; Tai, Y.; Treon, S.; Lin, B.; Schlossman, R.; Richardson, P.; et al. Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 2000, 96, 2943–2950. [Google Scholar] [CrossRef]
- Richardson, P.; Jagannath, S.; Hussein, M.; Berenson, J.; Singhal, S.; Irwin, D.; Williams, S.F.; Bensinger, W.; Badros, A.Z.; Vescio, R.; et al. Safety and efficacy of single-agent lenalidomide in patients with relapsed and refractory multiple myeloma. Blood 2009, 114, 772–778. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Zhu, Y.; Braggio, E.; Shi, C.-X.; Kortuem, M.K.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Wier, S.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, L.P.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef]
- Lagrue, K.; Carisey, A.; Morgan, D.J.; Chopra, R.; Davis, D.M. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood 2015, 126, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Kortuem, M.K.; Stewart, K.A. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leukemia Lymphoma 2012, 54, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Lacy, M.Q.; Hayman, S.R.; Gertz, M.A.; Dispenzieri, A.; Buadi, F.; Kumar, S.; Greipp, P.R.; Lust, J.A.; Russell, S.J.; Dingli, D.; et al. Pomalidomide (CC4047) Plus Low-Dose Dexamethasone As Therapy for Relapsed Multiple Myeloma. J. Clin. Oncol. 2009, 27, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Miguel, J.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066. [Google Scholar] [CrossRef]
- Richardson, P.G.; Siegel, D.S.; Vij, R.; Hofmeister, C.C.; Baz, R.; Jagannath, S.; Chen, C.; Lonial, S.; Jakubowiak, A.; Bahlis, N.; et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: A randomized phase 2 study. Blood 2014, 123, 1826–1832. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.-V.; Sonneveld, P.; et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2016, 127, 681–695. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Laubach, J.; Prada, P.C.; Richardson, P.; Longo, D. Daratumumab, Elotuzumab, and the Development of Therapeutic Monoclonal Antibodies in Multiple Myeloma. Clin. Pharmacol. Ther. 2017, 101, 81–88. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. New Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Durie, B.; Palumbo, A.; San-Miguel, J. Monoclonal antibodies in the treatment of multiple myeloma: Current status and future perspectives. Leukemia 2016, 30, 526–535. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Lonial, S. Novel Drug Combinations for the Management of Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leukemia 2014, 14, S71–S77. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. New Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. New Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. New Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. New Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Deckert, J.; Wetzel, M.-C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallée, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, A Novel Humanized CD38-Targeting Antibody, Demonstrates Potent Antitumor Activity in Models of Multiple Myeloma and Other CD38+ Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Attal, M.; Campana, F.; Le-Guennec, S.; Hui, A.-M.; Risse, M.-L.; Corzo, K.; Anderson, K.C. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Futur. Oncol. Lond. Engl. 2018, 14, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Goldschmidt, H.; Cavo, M.; Martin, T.G.; Paux, G.; Oprea, C.; Facon, T. Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM). J. Clin. Oncol. 2018, 36, TPS8055. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a Potential New Therapeutic Antibody Target for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Zonder, J.A.; Mohrbacher, A.F.; Singhal, S.; van Rhee, F.; Bensinger, W.I.; Ding, H.; Fry, J.; Afar, D.E.; Singhal, A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012, 120, 552–559. [Google Scholar] [CrossRef]
- Richardson, P.G.; Jagannath, S.; Moreau, P.; Jakubowiak, A.J.; Raab, M.S.; Facon, T.; Vij, R.; White, D.; Reece, D.E.; Benboubker, L.; et al. Elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: Final phase 2 results from the randomised, open-label, phase 1b–2 dose-escalation study. Lancet Haematol. 2015, 2, e516–e527. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. New Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncology 2018, 23, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Afifi, S.; Michael, A.; Azimi, M.; Rodriguez, M.; Lendvai, N.; Landgren, O. Role of Histone Deacetylase Inhibitors in Relapsed Refractory Multiple Myeloma: A Focus on Vorinostat and Panobinostat. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2015, 35, 1173–1188. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.; Elghandour, A.; Jedrzejczak, W.; Günther, A.; Nakorn, T.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hungria, V.T.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Nakorn, T.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by prior treatment. Blood 2016, 127, 713–721. [Google Scholar] [CrossRef]
- Hansen, V.L.; Coleman, M.; Elkins, S.; Letzer, J.P.; Levy, M.; Seneviratne, L.; Rine, J.; White, M.; Kuriakose, E.T. An Expanded Treatment Protocol of Panobinostat Plus Bortezomib and Dexamethasone in Patients With Previously Treated Myeloma. Clin. Lymphoma Myeloma Leukemia 2018, 18, 400–407.e1. [Google Scholar] [CrossRef]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I Study of Vorinostat in Combination with Bortezomib for Relapsed and Refractory Multiple Myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Siegel, D.S.; Lonial, S.; Qi, J.; Hajek, R.; Facon, T.; Rosinol, L.; Williams, C.; Blacklock, H.; Goldschmidt, H.; et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): A multicentre, randomised, double-blind study. Lancet Oncol. 2013, 14, 1129–1140. [Google Scholar] [CrossRef]
- Siegel, D.; Richardson, P.; Moreau, P.; Mitsiades, C.; Weber, D.; Houp, J.; Gause, C.; Vuocolo, S.; Eid, J.; Graef, T.; et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 2014, 4, e182. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. New Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Chen, C.; Siegel, D.; Gutierrez, M.; Jacoby, M.; Hofmeister, C.C.; Gabrail, N.; Baz, R.; Mau-Sorensen, M.; Berdeja, J.G.; Savona, M.; et al. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood 2018, 131, 855–863. [Google Scholar] [CrossRef]
- Syed, Y.Y. Selinexor: First Global Approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- Romero, D. Responses to selinexor in multiple myeloma. Nat. Rev. Clin. Oncol. 2019, 16, 661. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Richardson, P.G.; Moreau, P.; Anderson, K.C. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat. Rev. Clin. Oncol. 2015, 12, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-C.; Lin, S.-F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. Biomed. Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef]
- Pandey, M.; Amin, S.; Zangari, M.; Talamo, G. Drug Resistance in Multiple Myeloma: How to Cross the Border. Ann. Hematol. Oncol. 2015, 2, 01–07. [Google Scholar]
- Abraham, J.; Salama, N.N.; Azab, A. The role of P-glycoprotein in drug resistance in multiple myeloma. Leukemia Lymphoma 2014, 56, 26–33. [Google Scholar] [CrossRef]
- Franqui-Machin, R.; Wendlandt, E.B.; Janz, S.; Zhan, F.; Tricot, G. Cancer stem cells are the cause of drug resistance in multiple myeloma: Fact or fiction? Oncotarget 2015, 6, 40496–40506. [Google Scholar] [CrossRef]
- Sirohi, B.; Powles, R. Multiple myeloma. Lancet 2004, 363, 875–887. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, nrc2189. [Google Scholar] [CrossRef]
- Xavier, C.P.; Pesic, M.; Vasconcelos, M.H. Understanding Cancer Drug Resistance by Developing and Studying Resistant Cell Line Models. Curr. Cancer Drug Targets 2016, 16, 226–237. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Leleu, X.; Roussel, M.; Moreau, P.; Guerin-Charbonnel, C.; Caillot, D.; Marit, G.; Benboubker, L.; Voillat, L.; Mathiot, C.; et al. Bortezomib Plus Dexamethasone Induction Improves Outcome of Patients With t(4;14) Myeloma but Not Outcome of Patients With del(17p). J. Clin. Oncol. 2010, 28, 4630–4634. [Google Scholar] [CrossRef]
- Nemec, P.; Zemanova, Z.; Greslikova, H.; Michalova, K.; Filkova, H.; Tajtlova, J.; Kralova, D.; Kupska, R.; Smetana, J.; Krejci, M.; et al. Gain of 1q21 Is an Unfavorable Genetic Prognostic Factor for Multiple Myeloma Patients Treated with High-Dose Chemotherapy. Biol. Blood Marrow Transplant. 2010, 16, 548–554. [Google Scholar] [CrossRef]
- Sutlu, T.; Alici, E.; Jansson, M.; Wallblom, A.; Dilber, M.; Gahrton, G.; Nahi, H. The prognostic significance of 8p21 deletion in multiple myeloma. Brit. J. Haematol. 2009, 144, 266–268. [Google Scholar] [CrossRef]
- Sekiguchi, N.; Ootsubo, K.; Wagatsuma, M.; Midorikawa, K.; Nagata, A.; Noto, S.; Yamada, K.; Takezako, N. The impact of C-Myc gene-related aberrations in newly diagnosed myeloma with bortezomib/dexamethasone therapy. Int. J. Hematol. 2014, 99, 288–295. [Google Scholar] [CrossRef]
- Hao, M.; Zhang, L.; An, G.; Sui, W.; Yu, Z.; Zou, D.; Xu, Y.; Chang, H.; Qiu, L. Suppressing miRNA-15a/-16 expression by interleukin-6 enhances drug-resistance in myeloma cells. J. Hematol. Oncol. 2011, 4, 37. [Google Scholar] [CrossRef]
- Tian, Z.; Zhao, J.J.; Tai, Y.T.; Amin, S.B.; Hu, Y.; Berger, A.J.; Richardsonm, P.; Chauhan, D.; Anderson, K.C. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood 2012, 120, 3958–3967. [Google Scholar]
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; DePalma, L.; Chang, Y.; Peng, W.; Zhu, J.; Hawley, R.G. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol. 2013, 88, 265–272. [Google Scholar] [CrossRef]
- Besse, A.; Stolze, S.; Rasche, L.; Weinhold, N.; Morgan, G.; Kraus, M.; Bader, J.; Overkleeft, H.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef]
- Gouill, S.; Podar, K.; Harousseau, J.-L.; Anderson, K.C. Mcl-1 Regulation and Its Role in Multiple Myeloma. Cell Cycle 2004, 3, 1259–1262. [Google Scholar] [CrossRef]
- Zhang, B.; Gojo, I.; Fenton, R.G. Myeloid cell factor–1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885–1893. [Google Scholar] [CrossRef]
- Wuillème-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Gouill, L.S.; Avet-Loiseau, H.; Harousseau, J.-L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef]
- Qu, X.; Du, J.; Zhang, C.; Fu, W.; Xi, H.; Zou, J.; Hou, J. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PLoS ONE 2012, 7, e32215. [Google Scholar]
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97. [Google Scholar] [CrossRef]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omedè, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M.; et al. High XBP1 expression is a marker of better outcome in multiple myeloma patients treated with bortezomib. Haematologica 2014, 99, e14–e16. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef]
- Chatterjee, M.; Jain, S.; Stühmer, T.; Andrulis, M.; Ungethüm, U.; Kuban, R.-J.; Lorentz, H.; Bommert, K.; Topp, M.; Krämer, D.; et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90α and β in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 2007, 109, 720–728. [Google Scholar] [CrossRef]
- Khong, T.; Spencer, A. Targeting HSP 90 Induces Apoptosis and Inhibits Critical Survival and Proliferation Pathways in Multiple Myeloma. Mol. Cancer Ther. 2011, 10, 1909–1917. [Google Scholar] [CrossRef]
- Richardson, P.G.; Mitsiades, C.S.; Laubach, J.P.; Lonial, S.; Chanan-Khan, A.A.; Anderson, K.C. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Brit. J. Haematol. 2011, 152, 367–379. [Google Scholar] [CrossRef]
- Chatterjee, M.; Andrulis, M.; Stühmer, T.; Müller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef]
- Ishii, T.; Seike, T.; Nakashima, T.; Juliger, S.; Maharaj, L.; Soga, S.; Akinaga, S.; Cavenagh, J.; Joel, S.; Shiotsu, Y. Anti-tumor activity against multiple myeloma by combination of KW-2478, an Hsp90 inhibitor, with bortezomib. Blood Cancer J. 2012, 2, e68. [Google Scholar] [CrossRef][Green Version]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Kung, A.L.; Davies, F.E.; Morgan, G.; Akiyama, M.; Shringarpure, R.; Munshi, N.C.; et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood 2006, 107, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Saltarella, I.; Lamanuzzi, A.; Reale, A.; Vacca, A.; Ria, R. Identify multiple myeloma stem cells: Utopia? World J. Stem Cells 2015, 7, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, J.; Tolomelli, G.; Xu, H.; Xia, J.; Wang, H.; Zhou, W.; Zhou, Y.; Das, S.; Gu, Z.; et al. RARα2 expression confers myeloma stem cell features. Blood 2013, 122, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell–derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]
- Bianchi, G.; Kumar, S.; Ghobrial, I.M.; Roccaro, A.M.; Clinic, U.M. Cell Trafficking in Multiple Myeloma. Open J. Hematol. 2012, 3, 1. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.; Saha, M.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726. [Google Scholar] [CrossRef]
- Verbrugge, S.; Assaraf, Y.G.; Dijkmans, B.A.; Scheffer, G.L.; Al, M.; den Uyl, D.; Oerlemans, R.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; et al. Inactivating PSMB5 Mutations and P-Glycoprotein (Multidrug Resistance-Associated Protein/ATP-Binding Cassette B1) Mediate Resistance to Proteasome Inhibitors: Ex Vivo Efficacy of (Immuno)Proteasome Inhibitors in Mononuclear Blood Cells from Patients with Rheumatoid Arthritis. J. Pharmacol. Exp. Ther. 2012, 341, 174–182. [Google Scholar]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Kortüm, M.K.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- Derksen, P.W.; Tjin, E.; Meijer, H.P.; Klok, M.D.; Gillavry, H.D.; van Oers, M.H.; Lokhorst, H.M.; Bloem, A.C.; Clevers, H.; Nusse, R.; et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6122–6127. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Nijhof, I.; Groen, R.; Lokhorst, H.; van Kessel, B.; Bloem, A.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.; Klein, S.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Litzow, M.R.; Henderson, K.J.; Wier, S.A.; Ahmann, G.J.; Fonseca, R. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood 2005, 106, 2837–2840. [Google Scholar] [CrossRef]
- Greco, C.; D’Agnano, I.; Vitelli, G.; Vona, R.; Marino, M.; Mottolese, M.; Zuppi, C.; Capoluongo, E.; Ameglio, F. c-MYC deregulation is involved in melphalan resistance of multiple myeloma: role of PDGF-BB. Int. J. Immunopath. Pharmacol. 2006, 19, 67–79. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Gimsing, P.; Grønbæk, K. Aberrant microRNA expression in multiple myeloma. Eur. J. Haematol. 2013, 91, 95–105. [Google Scholar] [CrossRef]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef]
- Damiano, J.; Cress, A.; Hazlehurst, L.; Shtil, A.; Dalton, W. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Nass, J.; Efferth, T. Drug targets and resistance mechanisms in multiple myeloma. Cancer Drug Resist. 2018, 1, 87–117. [Google Scholar] [CrossRef]
- Fonseca, R.; Barlogie, B.; Bataille, R.; Bastard, C.; Bergsagel, L.P.; Chesi, M.; Davies, F.E.; Drach, J.; Greipp, P.R.; Kirsch, I.R.; et al. Genetics and Cytogenetics of Multiple Myeloma A Workshop Report. Cancer Res. 2004, 64, 1546–1558. [Google Scholar] [CrossRef]
- Rajan, A.; Rajkumar, S. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015, 5, e365. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, L.P.; Kuehl, M.W. Molecular Pathogenesis and a Consequent Classification of Multiple Myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef] [PubMed]
- Talley, P.J.; Chantry, A.D.; Buckle, C.H. Genetics in myeloma: genetic technologies and their application to screening approaches in myeloma. Brit. Med. Bull. 2015, 113, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Seidl, S.; Kaufmann, H.; Drach, J. New insights into the pathophysiology of multiple myeloma. Lancet Oncol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.; Munshi, N.C.; Anderson, K.C. Focus on multiple myeloma. Cancer Cell 2004, 6, 439–444. [Google Scholar] [CrossRef]
- Bergsagel, L.P.; Mateos, M.-V.; Gutierrez, N.C.; Rajkumar, V.S.; Miguel, J.F. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013, 121, 884–892. [Google Scholar] [CrossRef]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89. [Google Scholar] [CrossRef]
- Chesi, M.; Bergsagel, P.; Shonukan, O.; Martelli, M.; Brents, L.; Chen, T.; Schröck, E.; Ried, T.; Kuehl, W. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 1998, 91, 4457–4463. [Google Scholar] [CrossRef]
- Brito, J.; Walker, B.; Jenner, M.; Dickens, N.J.; Brown, N.; Ross, F.M.; Avramidou, A.; Irving, J.; Gonzalez, D.; Davies, F.E.; et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009, 94, 78–86. [Google Scholar] [CrossRef]
- Chesi, M.; Brents, L.A.; Ely, S.A.; Bais, C.; Robbiani, D.F.; Mesri, E.A.; Kuehl, M.W.; Bergsagel, L.P. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood 2001, 97, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.M.; Chiecchio, L.; Dagrada, G.; Protheroe, R.K.; Ockley, D.; Harrison, C.J.; Cross, N.C.; Szubert, A.J.; Drayson, M.T.; Morgan, G.J.; et al. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica 2010, 95, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Boersma-Vreugdenhil, G.R.; Kuipers, J.; Stralen, E.; Peeters, T.; Michaux, L.; Hagemeijer, A.; Pearson, P.L.; Clevers, H.C.; Bast, B.J. The recurrent translocation t(14;20)(q32;q12) in multiple myeloma results in aberrant expression of MAFB: A molecular and genetic analysis of the chromosomal breakpoint. Brit. J. Haematol. 2004, 126, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Hanamura, I.; Iida, S.; Akano, Y.; Hayami, Y.; Kato, M.; Miura, K.; Harada, S.; Banno, S.; Wakita, A.; Kiyoi, H.; et al. Ectopic Expression of MAFB Gene in Human Myeloma Cells Carrying (14;20)(q32;q11) Chromosomal Translocations. Jpn. J. Cancer Res. 2001, 92, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Audt, L. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Ngo, B.; Felthaus, J.; Hein, M.; Follo, M.; Wider, D.; Ihorst, G.; Engelhardt, M.; Wäsch, R. Monitoring bortezomib therapy in multiple myeloma: Screening of cyclin D1, D2, and D3 via reliable real-time polymerase chain reaction and association with clinico-pathological features and outcome. Leukemia Lymphoma 2010, 51, 1632–1642. [Google Scholar]
- Sewify, E.M.; Afifi, O.A.; Mosad, E.; Zaki, A.H.; Gammal, S.A. Cyclin D1 Amplification in Multiple Myeloma Is Associated With Multidrug Resistance Expression. Clin. Lymphoma Myeloma Leukemia. 2014, 14, 215–222. [Google Scholar] [CrossRef]
- Elnenaei, M.O.; Gruszka-Westwood, A.M.; A’Hernt, R.; Matutes, E.; Sirohi, B.; Powles, R.; Catovsky, D. Gene abnormalities in multiple myeloma; the relevance of TP53, MDM2, and CDKN2A. Haematologica 2003, 88, 529–537. [Google Scholar]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef]
- Reece, D.; Song, K.W.; Fu, T.; Roland, B.; Chang, H.; Horsman, D.E.; Mansoor, A.; Chen, C.; Masih-Khan, E.; Trieu, Y.; et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: Adverse effect of deletion 17p13. Blood 2009, 114, 522–525. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzalez, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosomes Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Kastritis, E.; Christoulas, D.; Migkou, M.; Gavriatopoulou, M.; Gkotzamanidou, M.; Iakovaki, M.; Matsouka, C.; Mparmparoussi, D.; Roussou, M.; et al. Treatment of patients with relapsed/refractory multiple myeloma with lenalidomide and dexamethasone with or without bortezomib: Prospective evaluation of the impact of cytogenetic abnormalities and of previous therapies. Leukemia 2010, 24, 1769. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Facon, T.; Grosbois, B.; Magrangeas, F.; Rapp, M.-J.; Harousseau, J.-L.; Minvielle, S.; Bataille, R.; du Myélome, I. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood 2002, 99, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Sagaster, V.; Ludwig, H.; Kaufmann, H.; Odelga, V.; Zojer, N.; Ackermann, J.; Küenburg, E.; Wieser, R.; Zielinski, C.; Drach, J. Bortezomib in relapsed multiple myeloma: Response rates and duration of response are independent of a chromosome 13q-deletion. Leukemia 2007, 21, 164–168. [Google Scholar] [CrossRef]
- Qazilbash, M.H.; Saliba, R.M.; Ahmed, B.; Parikh, G.; Mendoza, F.; Ashraf, N.; Hosing, C.; Flosser, T.; Weber, D.M.; Wang, M.; et al. Deletion of the Short Arm of Chromosome 1 (del 1p) is a Strong Predictor of Poor Outcome in Myeloma Patients Undergoing an Autotransplant. Biol. Blood Marrow Transplant. 2007, 13, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Walker, B.A.; Wardell, C.P.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gregory, W.M.; Jackson, G.H.; et al. Mapping of Chromosome 1p Deletions in Myeloma Identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as Being Genes in Regions Associated with Adverse Survival. Clin. Cancer Res. 2011, 17, 7776–7784. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Jung, S.-H.; Yang, D.-H.; Bae, S.-Y.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. Patterns of Relapse or Progression After Bortezomib-Based Salvage Therapy in Patients With Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leukemia 2014, 14, 389–394. [Google Scholar] [CrossRef]
- Duru, A.; Sutlu, T.; Wallblom, A.; Uttervall, K.; Lund, J.; Stellan, B.; Gahrton, G.; Nahi, H.; Alici, E. Deletion of Chromosomal Region 8p21 Confers Resistance to Bortezomib and Is Associated with Upregulated Decoy TRAIL Receptor Expression in Patients with Multiple Myeloma. PLoS ONE 2015, 10, e0138248. [Google Scholar] [CrossRef]
- Walker; Wardell, C.; Brioli, A.; Boyle, E.; Kaiser, M.; Begum, D.; Dahir, N.; Johnson, D.; Ross, F.; Davies, F.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191. [Google Scholar] [CrossRef]
- Chng, W.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.; Oken, M.; Kyle, R.; Henderson, K.; Wier, V.S.; Greipp, P.; et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Pichiorri, F.; Luca, L.; Aqeilan, R.I. MicroRNAs: New Players in Multiple Myeloma. Front. Genet. 2011, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Rocci, A.; Hofmeister, C.C.; Pichiorri, F. The potential of miRNAs as biomarkers for multiple myeloma. Expert Rev. Mol. Diagn. 2014, 14, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Sive, J.I.; Feber, A.; Smith, D.; Quinn, J.; Beck, S.; Yong, K. Global hypomethylation in myeloma is associated with poor prognosis. Brit. J. Haematol. 2016, 172, 473–475. [Google Scholar] [CrossRef]
- Bi, C.; Chng, W. MicroRNA: Important Player in the Pathobiology of Multiple Myeloma. Biomed. Res. Int. 2014, 2014, 521586. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.-S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 2009, 113, 6669–6680. [Google Scholar] [CrossRef]
- Wang, X.; Li, C.; Ju, S.; Wang, Y.; Wang, H.; Zhong, R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leukemia Lymphoma 2011, 52, 1991–1998. [Google Scholar] [CrossRef]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6–dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef]
- Leone, E.; Morelli, E.; Martino, M.T.; Amodio, N.; Foresta, U.; Gullà, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 Inhibits In Vitro and In Vivo Multiple Myeloma Cell Growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef]
- Munker, R.; Liu, C.-G.; Taccioli, C.; Alder, H.; Heerema, N. MicroRNA Profiles of Drug-Resistant Myeloma Cell Lines. Acta Haematol. 2010, 123, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Wang, H.; Leng, Y.; Li, Z.-L.; Yang, Y.-F.; Xiao, F.-J.; Li, Q.-F.; Chen, X.-Q.; Wang, L.-S. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem. Biophys. Res. Commun. 2011, 414, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Martino, D.M.; Foresta, U.; Leone, E.; Lionetti, M.; Leotta, M.; Gullà, A.; Pitari, M.; Conforti, F.; Rossi, M.; et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012, 3, e436. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. MECHANISMS OF CANCER DRUG RESISTANCE. Medicine 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar]
- Dalton, W. Detection of multidrug resistance gene expression in multiple myeloma. Leukemia 1997, 11, 1166–1169. [Google Scholar] [CrossRef]
- Nooter, K.; Stoter, G. Molecular Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Pathol. Res. Pract. 1996, 192, 768–780. [Google Scholar] [CrossRef]
- Grogan, T.; Spier, C.; Salmon, S.; Matzner, M.; Rybski, J.; Weinstein, R.; Scheper, R.; Dalton, W. P-glycoprotein expression in human plasma cell myeloma: Correlation with prior chemotherapy. Blood 1993, 81, 490–495. [Google Scholar] [CrossRef]
- Chen, N.; Weiss, D.; Reyes, J.; Liu, L.; Kasserra, C.; Wang, X.; Zhou, S.; Kumar, G.; Weiss, L.; Palmisano, M. No clinically significant drug interactions between lenalidomide and P-glycoprotein substrates and inhibitors: Results from controlled phase I studies in healthy volunteers. Cancer Chemoth. Pharm. 2014, 73, 1031–1039. [Google Scholar] [CrossRef]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemoth. Pharm. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting Apoptosis Pathways in Cancer Therapy. Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.H.; Ma, M.H.; Vescio, R.A.; Berenson, J.R. Overcoming Drug Resistance in Multiple Myeloma: The Emergence of Therapeutic Approaches to Induce Apoptosis. J. Clin. Oncol. 2003, 21, 4239–4247. [Google Scholar] [CrossRef] [PubMed]
- Lentzsch, S.; Chatterjee, M.; Gries, M.; Bommert, K.; Gollasch, H.; Dörken, B.; Bargou, R. PI3-K/AKT/FKHR and MAPK signaling cascades are redundantly stimulated by a variety of cytokines and contribute independently to proliferation and survival of multiple myeloma cells. Leukemia 2004, 18, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Ge, N.; Rudikoff, S. Insulin-like growth factor I is a dual effector of multiple myeloma cell growth. Blood 2000, 96, 2856–2861. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gardner, A.; Lichtenstein, A. The phosphatidylinositol 3-kinase/AKT kinase pathway in multiple myeloma plasma cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000, 60, 6763–6770. [Google Scholar] [PubMed]
- Mitsiades, C.S.; Treon, S.P.; Mitsiades, N.; Shima, Y.; Richardson, P.; Schlossman, R.; Hideshima, T.; Anderson, K.C. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: Therapeutic applications. Blood 2001, 98, 795–804. [Google Scholar] [CrossRef]
- Balsas, P.; López-Royuela, N.; Galán-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef]
- Jourdan, M.; Veyrune, J.-L.; Vos, J.; Redal, N.; Couderc, G.; Klein, B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene 2003, 22, 2950–2959. [Google Scholar] [CrossRef]
- Puthier, D.; Bataille, R.; Amiot, M. IL-6 up-regulates Mcl-1 in human myeloma cells through JAK / STAT rather than Ras / MAP kinase pathway. Eur. J. Immunol. 1999, 29, 3945–3950. [Google Scholar] [CrossRef]
- Gouill, S.; Podar, K.; Amiot, M.; Hideshima, T.; Chauhan, D.; Ishitsuka, K.; Kumar, S.; Raje, N.; Richardson, P.G.; Harousseau, J.-L.; et al. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood 2004, 104, 2886–2892. [Google Scholar] [CrossRef]
- Spets, H.; Strömberg, T.; Georgii-Hemming, P.; Siljason, J.; Nilsson, K.; Jernberg-Wiklund, H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells. Eur. J. Haematol. 2002, 69, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dong, L.; Castro, A.; Palombella, V.; et al. NF-κB as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Vincenz, L.; Jäger, R.; O’Dwyer, M.; Samali, A. Endoplasmic Reticulum Stress and the Unfolded Protein Response: Targeting the Achilles Heel of Multiple Myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.-S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Montanari, F.; Lu, M.; Marcus, S.; Saran, A.; Malankar, A.; Mazumder, A. A Phase II Trial of Chloroquine in Combination with Bortezomib and Cyclophosphamide in Patients with Relapsed and Refractory Multiple Myeloma. Blood 2014, 124, 5775. [Google Scholar] [CrossRef]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de Miguel, D.; Ramírez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef]
- Huston, A.; Leleu, X.; Jia, X.; Moreau, A.-S.; Ngo, H.T.; Runnels, J.; Anderson, J.; Alsayed, Y.; Roccaro, A.; Vallet, S.; et al. Targeting Akt and Heat Shock Protein 90 Produces Synergistic Multiple Myeloma Cell Cytotoxicity in the Bone Marrow Microenvironment. Clin. Cancer Res. 2008, 14, 865–874. [Google Scholar] [CrossRef][Green Version]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drugs Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Bergsagel, D.; McCulloch, E. Mouse myeloma tumor stem cells: a primary cell culture assay. J. Natl. Cancer Inst. 1971, 46, 411–422. [Google Scholar] [PubMed]
- Matsui, W.; Wang, Q.; Barber, J.P.; Brennan, S.; Smith, D.B.; Borrello, I.; McNiece, I.; Lin, L.; Ambinder, R.F.; Peacock, C.; et al. Clonogenic Multiple Myeloma Progenitors, Stem Cell Properties, and Drug Resistance. Cancer Res. 2008, 68, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Matsui, W.; Huff, C.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, D.B.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Matsui, W. Cancer stem cells in multiple myeloma. Cancer Lett. 2009, 277, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Park, C.Y.; Medeiros, B.C.; Weissman, I.L. CD19-CD45 low/-CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012, 26, 2530–2537. [Google Scholar] [CrossRef]
- Gao, M.; Kong, Y.; Yang, G.; Gao, L.; Shi, J. Multiple myeloma cancer stem cells. Oncotarget 2016, 7, 35466–35477. [Google Scholar] [CrossRef]
- Gao, M.; Bai, H.; Jethava, Y.; Wu, Y.; Zhu, Y.; Yang, Y.; Via, J.; Cao, H.; Fraqui-Machin, R.; Nadiminti, K.; et al. Identification and Characterization of Tumor-Initiating Cells in Multiple Myeloma. J. Natl. Cancer Inst. 2019, 112, djz159. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef]
- Jundt, F.; Pröbsting, K.; Anagnostopoulos, I.; Muehlinghaus, G.; Chatterjee, M.; Mathas, S.; Bargou, R.C.; Manz, R.; Stein, H.; Dörken, B. Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood 2004, 103, 3511–3515. [Google Scholar] [CrossRef]
- Kellner, J.; Liu, B.; Kang, Y.; Li, Z. Fact or fiction - identifying the elusive multiple myeloma stem cell. J. Hematol. Oncol. 2013, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.; Nuñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Burger, R.; Gouill, S.; Tai, Y.-T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Favata, M.; Kelley, J.A.; Caulder, E.; Thomas, B.; Wen, X.; Sparks, R.B.; Arvanitis, A.; Rogers, J.D.; Combs, A.P.; et al. INCB16562, a JAK1/2 Selective Inhibitor, Is Efficacious against Multiple Myeloma Cells and Reverses the Protective Effects of Cytokine and Stromal Cell Support. Neoplasia 2010, 12, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Hunsucker, S.A.; Magarotto, V.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Weber, D.M.; Thomas, S.K.; Shah, J.J.; Voorhees, P.M.; Xie, H.; et al. Blockade of interleukin-6 signalling with siltuximab enhances melphalan cytotoxicity in preclinical models of multiple myeloma. Brit. J. Haematol. 2011, 152, 579–592. [Google Scholar] [CrossRef]
- Monaghan, K.; Khong, T.; Burns, C.; Spencer, A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 2011, 25, 1891–1899. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Chen, Q.; Kuhn, D.J.; Small, G.W.; Hunsucker, S.A.; Strader, J.S.; Corringham, R.E.; Zaki, M.H.; Nemeth, J.A.; Orlowski, R.Z. Inhibition of Interleukin-6 Signaling with CNTO 328 Enhances the Activity of Bortezomib in Preclinical Models of Multiple Myeloma. Clin. Cancer Res. 2007, 13, 6469–6478. [Google Scholar] [CrossRef]
- Lin, L.; Benson, D.M.; DeAngelis, S.; Bakan, C.E.; Li, P.; Li, C.; Lin, J. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 2012, 130, 1459–1469. [Google Scholar] [CrossRef]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.; Libermann, T.; Anderson, K. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996, 87, 1104–1112. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.; Elliott, P.; Adams, J.; Anderson, K. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Keifer, J.A.; Guttridge, D.C.; Ashburner, B.P.; Baldwin, A.S. Inhibition of NF-κB Activity by Thalidomide through Suppression of IκB Kinase Activity. J. Biol. Chem. 2001, 276, 22382–22387. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Yoon, S.-S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tai, Y.-T.; Davies, F.E.; Lentzsch, S.; Sattler, M.; Hideshima, T.; Lin, B.K.; Gupta, D.; Shima, Y.; Chauhan, D.; et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Podar, K.; Gupta, D.; Tai, Y.-T.; Li, S.; Weller, E.; Hideshima, T.; Lentzsch, S.; Davies, F.; Li, C.; et al. The vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002, 62, 5019–5026. [Google Scholar] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.; Vergara, D.; et al. A HGF/cMET Autocrine Loop Is Operative in Multiple Myeloma Bone Marrow Endothelial Cells and May Represent a Novel Therapeutic Target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; Bruyne, E.; Valckenborgh, E.; Lahoutte, T.; Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- De Luca, L.; Laurenzana, I.; Trino, S.; Lamorte, D.; Caivano, A.; Musto, P. An update on extracellular vesicles in multiple myeloma: A focus on their role in cell-to-cell crosstalk and as potential liquid biopsy biomarkers. Expert Rev. Mol. Diagn. 2019, 19, 249–258. [Google Scholar] [CrossRef]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.J.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promote Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Vasconcelos, M.H.; Caires, H.R.; Abols, A.; Xavier, C.P.R.; Line, A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drugs Resist. Updat. 2019, 47, 1000647. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Luca, A.; Sousa, D.; Seca, H.J.; Meleady, O.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug resistant tumour cells shed more microvesicle-like EVs and less exosomes than their drug-sensitive counterpart cells. Biochim. Biophys. Acta 2019, 6, 1023–2017. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Agents | Mechanism of Action | Type of Resistance | Mechanism of Resistance |
|---|---|---|---|
| Proteasome Inhibitors (bortezomib, carfilzomib and ixazomib) | Inhibition of proteasome activity; inhibition of NF-κB; induction of apoptosis by activating caspase-8 and caspase-9; upregulation NOXA; down-regulation of adhesion molecules [52,53,55,61]. | Genetics and Genomics | Mutations of gene TP53; mutation of gene MAF, t14;16) and t(14;20); point mutations of the gene PSMB5 with overexpression of B5 subunit; upregulation of the proteasomal system; overexpression of the gene MYC; 1q21 gain; modification or loss of 8p21 [122,123,124,125]. |
| Epigenetics | downregulation of miR-15a; downregulation of mir-33b [126,127]. | ||
| Abnormal Drug Transport | Upregulation of P-gp (mainly for carfilzomib) [128,129]. | ||
| Escape from apoptosis, autophagy and dysregulated intracellular signaling pathways | Upregulation of pro-survival proteins (Mcl-1, Bcl-2, Bcl-XL); constitutive activation of the NF-κB pathway; Activation of the aggresome and autophagy pathway; Low levels of the UPR transcription factor XBP1 and autophagy-inducer activating transcription factor 4; increase in heat shock proteins (Grp78, Hsp90, Hsp70, Hsp8) [130,131,132,133,134,135,136,137,138,139,140,141,142]. | ||
| Persistence of Cancer Stem Cells | Stem cell-like phenotype with increased levels of multidrug transporters (ABCG2/BCRP) and ALDH1A1 enzymatic activity; Activation of Hedgehog, Wnt and Notch pathways; upregulation of BTK receptors and RARa2 [118,143,144]. | ||
| Microenvironment | Proliferation and cell survival signaling such as IL6/JAK/STAT3, MAPK, PI3/AKT, IGF-1; Increase production of VEGF leading to angiogenesis, cell proliferation and migration; Increase of pro-inflammatory TNF-α; Increase of cell adhesion molecules (LFA1, VLA4, ICAM1, VCAM1); Activation of SDF1/CXCR4 axis; Increase expression of MARCKS in adhesion and metastatic spread; EVs cargo and cell communication [126,145,146,147,148]. | ||
| Immunomodulatory agents (thalidomide, lenalidomide, pomalidomide) | Interaction with BM microenvironment with down-regulation of adhesion molecules; targeting the cereblon and downstream targets; regulation of pro and anti-inflammatory cytokines; regulation of T cell and NK cells activity; anti-angiogenesis; induction of apoptosis by activating caspase 8 and 9 [64,65,66,67,68,69,70,71]. | Reduced target expression | Mutations in cereblon and genes in the cereblon-pathway (IFKF1 and KPNA2); Mutations in Ras/Raf pathway (KRAS G12D and BRAF V600E) [70,149,150,151]. |
| Genetics and Genomics | Mutations in cereblon and genes in the cereblon-pathway (IFKF1 and KPNA2) [151]. | ||
| Persistence of Cancer Stem Cells | Stem cell-like phenotype with increased levels of multidrug transporters (ABCG2/BCRP) and ALDH1A1 enzymatic activity; Activation of Hedgehog, Wnt and Notch pathways; upregulation of BTK receptors and RARa2 [118,143,144]. | ||
| Microenvironment | Increase of cell adhesion molecules (CD44 thought the Wnt/B-catenin signaling) [152]. | ||
| Monoclonal antibodies (daratumumab, elotuzumab, isatuximab) | Antibody-dependent cellular cytotoxicity; complement-dependent cytotoxicity; macrophage-mediated phagocytosis; FCyR-mediated crosslinking; modulation of target antigen enzymatic activity; regulation of Tregs and stimulation of T cell and NK activity [79,81,85,91,92,96]. | Reduced target expression | Reduced expression of CD38 and SLAM7 [153]. |
| Complement resistance | Increased expression of CD46, CD56 and CD59 blocking anti-body-induced CDC [153,154]. | ||
| Microenvironment | Competition by the extracellular soluble forms of CD38 and SLAM7 [78]. | ||
| Neutralization | Anti-idiotype antibodies neutralizing the activity of the therapeutic monoclonal antibodies [99]. | ||
| Histone deacetylase inhibitors (panobinostat, vorinostat) | Activation of tumour suppressor genes; synergetic activity with proteasomal and aggresomal protein degradation; upregulation p21 [101,102]. | Escape from apoptosis, autophagy activation and dysregulated intracellular signaling pathways | Abnormal regulation of actin cytoskeleton and abnormal protein processing in endoplasmic reticulum (activation of PI3/AKT, MEK/ERK and FAK signaling pathway) [137,138,139,140] |
| Exportin 1 inhibitors (selinexor) | Nuclear retention and activation of tumour suppressor genes, inhibition of NF-κB; reduction of oncoprotein mRNAs translation [109,110]. | - | - |
| Alkylating agents (melphalan, cyclophosphamide) and Anthracyclines (doxorubicin) | DNA damage; topoisomerase II inhibition | Genetics, Genomics and Epigenetics | Overexpression of the gene MYC; upregulation of miR-21; downregulation of miR-15a [126,155,156,157]. |
| Abnormal Drug Transport | Upregulation of P-gp; increased ABCG2 expression [117,158]. | ||
| Persistence of Cancer Stem Cells | Stem cell-like phenotype with increased levels of multidrug transporters (ABCG2/BCRP) and ALDH1A1 enzymatic activity; Activation of Hedgehog, Wnt and Notch pathways; upregulation of BTK receptors and RARa2 [117,118,144,152,158]. | ||
| Microenvironment | Increase of cell adhesion molecules (VLA4) [159]. | ||
| Corticosteroids (dexamethasone, prednisolone, methylprednisolone) | Induction of apoptosis | Reduced target expression | Functional defect of the glucocorticoid receptor [114,115,116,117]. |
| Genetics, Genomics and Epigenetics | Overexpression of the gene MYC and FGFR3; epigenetic inactivation of RASD1 [114,115,116,117]. | ||
| Microenvironment | Increase secretion of pro-survival cytokines [114,115,116,117] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. https://doi.org/10.3390/cancers12020407
Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers. 2020; 12(2):407. https://doi.org/10.3390/cancers12020407
Chicago/Turabian StylePinto, Vanessa, Rui Bergantim, Hugo R. Caires, Hugo Seca, José E. Guimarães, and M. Helena Vasconcelos. 2020. "Multiple Myeloma: Available Therapies and Causes of Drug Resistance" Cancers 12, no. 2: 407. https://doi.org/10.3390/cancers12020407
APA StylePinto, V., Bergantim, R., Caires, H. R., Seca, H., Guimarães, J. E., & Vasconcelos, M. H. (2020). Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers, 12(2), 407. https://doi.org/10.3390/cancers12020407