Metabolism of Estrogens: Turnover Differs between Platinum-Sensitive and -Resistant High-Grade Serous Ovarian Cancer Cells


 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Characterization of the Investigated HGSOC Cell Lines
2.2. DHEA Metabolism by Platinum-Sensitive and -Resistant HGSOC Cells
2.3. E1 Metabolism by Platinum-Sensitive and -Resistant HGSOC Cells
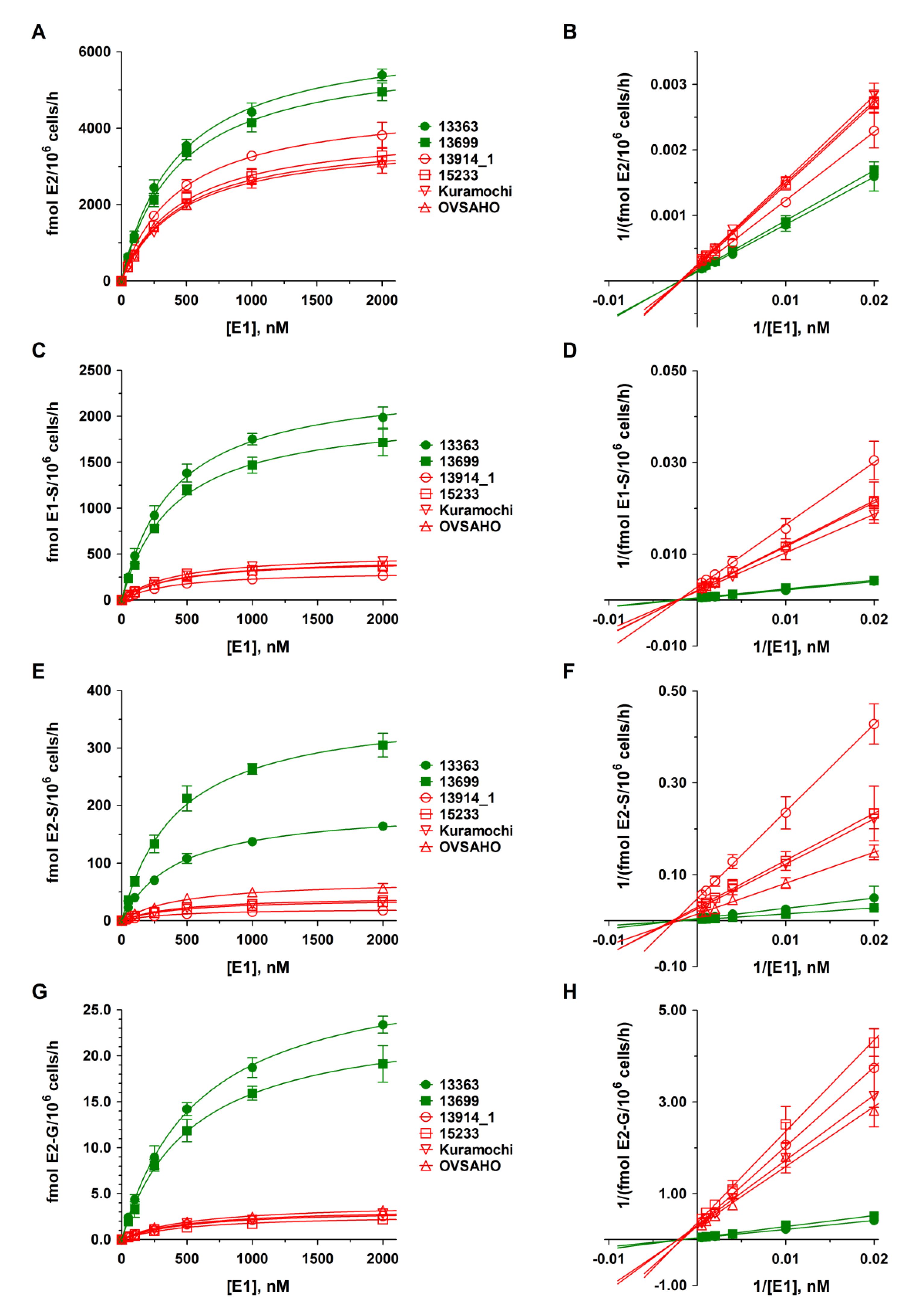
2.4. Kinetics of DHEA and E1 Metabolism in HGSOC Cells
2.5. Proliferation of Platinum-Sensitive and -Resistant HGSOC Cell Lines
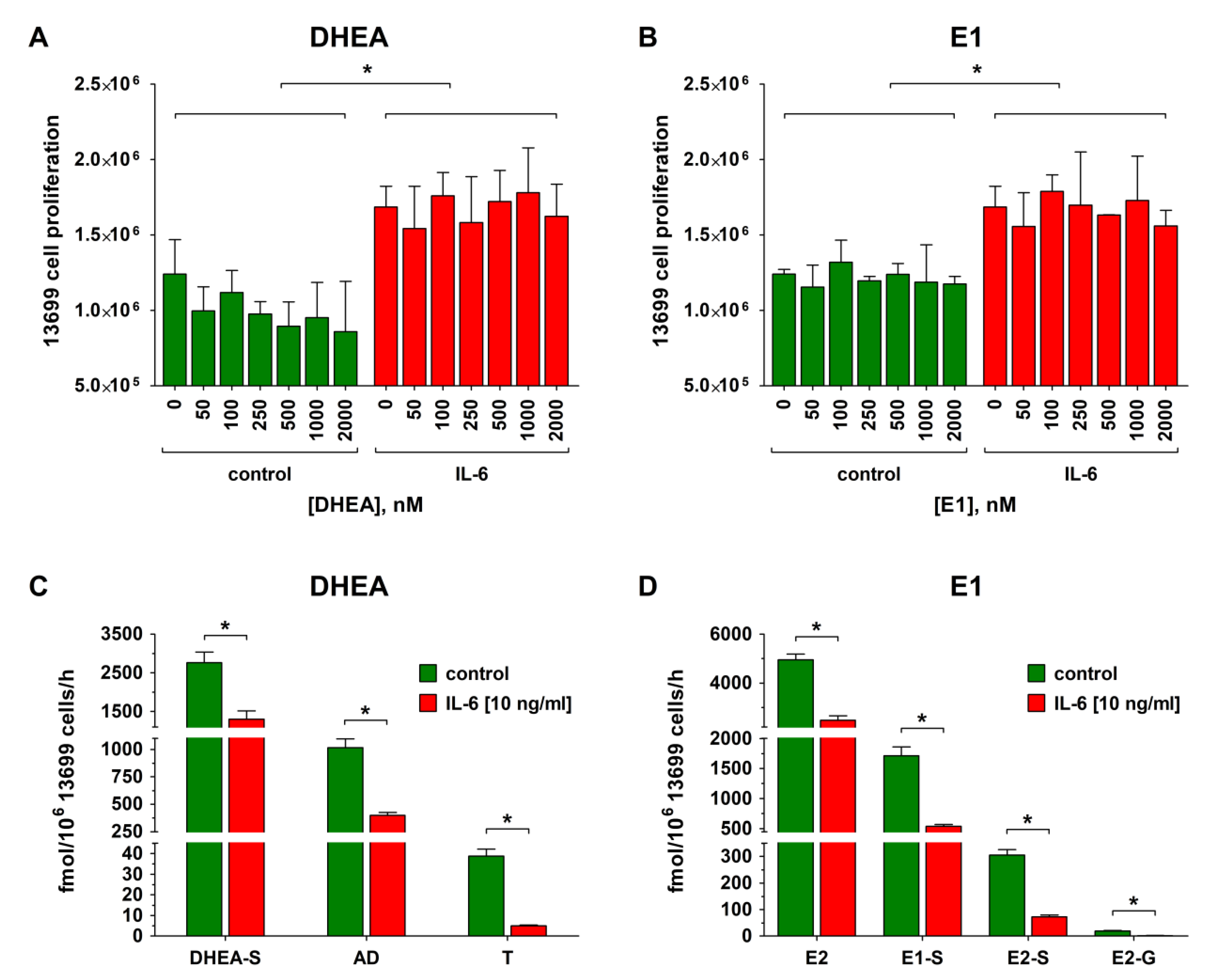
2.6. Effect of IL-6 on Proliferation, Metabolism, and Carboplatin Resistance of Platinum-Sensitive HGSOC Cells
2.7. Effect of Tocilizumab (TCZ) Treatment on Proliferation, Metabolism, and Carboplatin Resistance of Platinum-Resistant HGSOC Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Gene Expression Analyses and Identification of Gene Mutations
4.4. Carboplatin Resistance
4.5. Metabolism of Steroid Hormones by Platinum-Sensitive and -Resistant HGSOC Cells
4.6. LC-HRMS Assay for Steroid Quantification
4.7. IL-6 Determination in the Cellular Supernatants
4.8. Impact of IL-6 and TCZ on Metabolism and Progression of HGSOC Cells
4.9. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 17β-HSD | 17β-hydroxysteroid-dehydrogenase |
| 3β-HSD | 3β-hydroxysteroid-dehydrogenase |
| AD | 4-androstene-3,17-dione |
| CYP | cytochrome P450 |
| DHEA | dehydroepiandrosterone |
| DHEA-S | dehydroepiandrosterone-3-sulfate |
| DMEM/F-12 | Dulbecco’s modified Eagle medium F-12 |
| DPBS | Dulbecco’s phosphate buffered saline |
| E1 | estrone |
| E1-S | estrone-3-sulfate |
| E2 | 17β-estradiol |
| E2-G | 17β-estradiol-3-O-(β-d-glucuronide) |
| E2-S | 17β-estradiol-3-sulfate |
| E3 | estriol |
| EOC | epithelial ovarian cancer |
| ERα | estrogen receptor alpha |
| ESR1 | estrogen receptor alpha gene |
| ESR2 | estrogen receptor beta gene |
| ESRRG | estrogen-related receptor gamma gene |
| FBS | fetal bovine serum |
| HGSOC | high grade serous ovarian cancer |
| IL-6 | interleukin-6 |
| IL-6R | interleukin-6 receptor |
| Km | Michaelis constant |
| LC-HRMS | liquid chromatography high-resolution mass spectrometry |
| LLOQ | lower limit of quantification |
| PARP | poly(ADP-ribose)-polymerase 1 |
| PXR | pregnane X receptor |
| STR | short tandem repeats |
| SULT | sulfotransferase |
| T | testosterone |
| TCZ | tocilizumab |
| UGT | uridine 5’-diphospho-glucuronosyltransferase |
| VEGF | vascular endothelial growth factor |
| Vmax | maximum reaction velocity |
References
- Longuespée, R.; Boyon, C.; Desmons, A.; Vinatier, D.; Leblanc, E.; Farré, I.; Wisztorski, M.; Ly, K.; D’Anjou, F.; Day, R.; et al. Ovarian cancer molecular pathology. Cancer Metastasis Rev. 2012, 31, 713–732. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Makar, A.P. Hormone therapy in epithelial ovarian cancer. Endocr.-Relat. Cancer 2000, 7, 85–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rainczuk, A.; Rao, J.R.; Gathercole, J.L.; Fairweather, N.J.; Chu, S.; Masadah, R.; Jobling, T.W.; Deb-Choudhury, S.; Dyer, J.; Stephens, A.N. Evidence for the antagonistic form of CXC-motif chemokine CXCL10 in serous epithelial ovarian tumours. Int. J. Cancer 2014, 134, 530–541. [Google Scholar] [CrossRef]
- Kroeger, P.T.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Angioli, R.; Palaia, I.; Zullo, M.A.; Muzii, L.; Manci, N.; Calcagno, M.; Panici, P.B. Diagnostic open laparoscopy in the management of advanced ovarian cancer. Gynecol. Oncol. 2016, 100, 455–461. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Ivy, S.P.; Liu, J.F.; Lee, J.M.; Matulonis, U.A.; Kohn, E.C. Cediranib, a pan-VEGFR inhibitor, and olaparib, a PARP inhibitor, in combination therapy for high grade serous ovarian cancer. Expert Opin. Investig. Drugs 2016, 25, 597–611. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Karam, A.; Ledermann, J.A.; Kim, J.W.; Sehouli, J.; Lu, K.; Gourley, C.; Katsumata, N.; Burger, R.A.; Nam, B.H.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: First-line interventions. Ann. Oncol. 2017, 28, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, F.; Thalhammer, T. Estrogen biosynthesis and action in ovarian cancer. Front. Endocrinol. (Lausanne) 2014, 5, 192. [Google Scholar] [CrossRef] [PubMed]
- Moyle-Heyrman, G.; Schipma, M.J.; Dean, M.; Davis, D.A.; Burdette, J.E. Genome-wide transcriptional regulation of estrogen receptor targets in fallopian tube cells and the role of selective estrogen receptor modulators. J. Ovarian Res. 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Stanley, B.; Hollis, R.L.; Nunes, H.; Towler, J.D.; Yan, X.; Rye, T.; Dawson, C.; Mackean, M.J.; Nussey, F.; Churchman, M.; et al. Endocrine treatment of high grade serous ovarian carcinoma; quantification of efficacy and identification of response predictors. Gynecol. Oncol. 2019, 152, 278–285. [Google Scholar] [CrossRef]
- Paleari, L.; Gandini, S.; Provinciali, N.; Puntoni, M.; Colombo, N.; DeCensi, A. Clinical benefit and risk of death with endocrine therapy in ovarian cancer: A comprehensive review and meta-analysis. Gynecol. Oncol. 2017, 146, 504–513. [Google Scholar] [CrossRef]
- Stasenko, M.; Plegue, M.; Sciallis, A.P.; McLean, K. Clinical response to antiestrogen therapy in platinum-resistant ovarian cancer patients and the role of tumor estrogen receptor expression status. Int. J. Gynecol. Cancer 2015, 25, 222–228. [Google Scholar] [CrossRef]
- Ren, X.; Wu, X.; Hillier, S.G.; Fegan, K.S.; Critchley, H.O.; Mason, J.I.; Sarvi, S.; Harlow, C.R. Local estrogen metabolism in epithelial ovarian cancer suggests novel targets for therapy. J. Steroid Biochem. Mol. Biol. 2015, 150, 54–63. [Google Scholar] [CrossRef]
- Mungenast, F.; Aust, S.; Vergote, I.; Vanderstichele, A.; Sehouli, J.; Braicu, E.; Mahner, S.; Castillo-Tong, D.C.; Zeillinger, R.; Thalhammer, T. Clinical significance of the estrogen-modifying enzymes steroid sulfatase and estrogen sulfotransferase in epithelial ovarian cancer. Oncol. Lett. 2017, 13, 4047–4054. [Google Scholar] [CrossRef]
- Poschner, S.; Zehl, M.; Maier-Salamon, A.; Jäger, W. Simultaneous quantification of estrogens, their precursors and conjugated metabolites in human breast cancer cells by LC-HRMS without derivatization. J. Pharm. Biomed. Anal. 2017, 138, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, X.L.; Qu, Y.; Wu, J.; Zhu, Y.Q.; Sun, W.J.; Li, L.Z. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010, 295, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, C.; Rouzier, R.; Geyl, C.; Cortez, A.; Castela, M.; Lis, R.; Daraï, E.; Touboul, C. Predictive markers of chemoresistance in advanced stages epithelial ovarian carcinoma. Gynecol. Oncol. 2015, 136, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Penson, R.T.; Kronish, K.; Duan, Z.; Feller, A.J.; Stark, P.; Cook, S.E.; Duska, L.R.; Fuller, A.F.; Goodman, A.K.; Nikrui, N.; et al. Cytokines IL-1beta, IL-2, IL-6, IL-8, MCP-1, GM-CSF and TNFalpha in patients with epithelial ovarian cancer and their relationship to treatment with paclitaxel. Int. J. Gynecol. Cancer 2000, 10, 33–41. [Google Scholar] [CrossRef]
- Kreuzinger, C.; Gamperl, M.; Wolf, A.; Heinze, G.; Geroldinger, A.; Lambrechts, D.; Boeckx, B.; Smeets, D.; Horvat, R.; Aust, S.; et al. Molecular characterization of 7 new established cell lines from high grade serous ovarian cancer. Cancer Lett. 2015, 362, 218–228. [Google Scholar] [CrossRef]
- Haley, J.; Tomar, S.; Pulliam, N.; Xiong, S.; Perkins, S.M.; Karpf, A.R.; Mitra, S.; Nephew, K.P.; Mitra, A.K. Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget 2016, 7, 32810–32820. [Google Scholar] [CrossRef]
- He, Y.; Zhu, Q.; Chen, M.; Huang, Q.; Wang, W.; Li, Q.; Huang, Y.; Di, W. The changing 50% inhibitory concentration (IC50) of cisplatin: A pilot study on the artifacts of the MTT assay and the precise measurement of density-dependent chemoresistance in ovarian cancer. Oncotarget 2016, 7, 70803–70821. [Google Scholar] [CrossRef]
- Lindl, T.; Lewandowski, B.; Sheyrogg, S.; Staudte, A. Evaluation of the In Vitro Cytotoxicities of 50 Chemicals by using an Electronic Current Exclusion Method versus the Neutral Red Uptake and MTT Assays. ATLA 2005, 33, 591–601. [Google Scholar]
- Murias, M.; Miksits, M.; Aust, S.; Spatzenegger, M.; Thalhammer, T.; Szekeres, T.; Jaeger, W. Metabolism of resveratrol in breast cancer cell lines: Impact of sulfotransferase 1A1 expression on cell growth inhibition. Cancer Lett. 2008, 261, 172–182. [Google Scholar] [CrossRef]
- Imai, A.; Ohno, T.; Takahashi, K.; Furui, T.; Tamaya, T. Lack of evidence for aromatase expression in human ovarian epithelial carcinoma. Ann. Clin. Biochem. 1994, 31, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sasano, H.; Harada, N. Intratumoral aromatase in human breast, endometrial, and ovarian malignancies. Endocr. Rev. 1998, 19, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Blanco, L.Z., Jr.; Kuhn, E.; Morrison, J.C.; Bahadirli-Talbott, A.; Smith-Sehdev, A.; Kurman, R.J. Steroid hormone synthesis by the ovarian stroma surrounding epithelial ovarian tumors: A potential mechanism in ovarian tumorigenesis. Mod. Pathol. 2017, 30, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann-Schwarz, V.; Knipprath Mészaros, A.; Stadlmann, S.; Jacob, F.; Schoetzau, A.; Russell, K.; Friedlander, M.; Singer, G.; Vetter, M. Letrozole may be a valuable maintenance treatment in high-grade serous ovarian cancer patients. Gynecol. Oncol. 2018, 148, 79–85. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Q.; Miao, C.; Dongol, S.; Li, Y.; Jin, C.; Dong, R.; Li, Y.; Yang, X.; Kong, B. CCNG1 (Cyclin G1) regulation by mutant-P53 via induction of Notch3 expression promotes high-grade serous ovarian cancer (HGSOC) tumorigenesis and progression. Cancer Med. 2019, 8, 351–362. [Google Scholar] [CrossRef]
- Browning, L.; Patel, M.R.; Bring Horvath, E.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef]
- Strasser, S.I.; Mashford, M.L.; Desmond, P.V. Regulation of uridine diphosphate glucuronosyltransferase during the acute-phase response. J. Gastroenterol. Hepatol. 1998, 13, 88–94. [Google Scholar] [CrossRef]
- Simon, F.; Garcia, J.; Guyot, L.; Guitton, J.; Vilchez, G.; Bardel, C.; Chenel, M.; Tod, M.; Payen, L. Impact of Interleukin-6 on Drug-Metabolizing Enzymes and Transporters in Intestinal Cells. AAPS J. 2019, 22, 16. [Google Scholar] [CrossRef]
- Kim, S.; Östör, A.J.; Nisar, M.K. Interleukin-6 and cytochrome-P450, reason for concern? Rheumatol. Int. 2012, 32, 2601–2604. [Google Scholar] [CrossRef]
- Ning, R.; Zhan, Y.; He, S.; Hu, J.; Zhu, Z.; Hu, G.; Yan, B.; Yang, J.; Liu, W. Interleukin-6 Induces DEC1, Promotes DEC1 Interaction with RXRα and Suppresses the Expression of PXR, CAR and Their Target Genes. Front. Pharmacol. 2017, 8, 866. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Abualsunun, W.A.; Sahin, C.; Cummins, C.L.; Piquette-Miller, M. Essential role of STAT-3 dependent NF-κB activation on IL-6-mediated downregulation of hepatic transporters. Eur. J. Pharm. Sci. 2019, 143, 105151. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, H.; Momeny, M.; Ghaffari, S.H.; Parsanejad, N.; Poursheikhani, A.; Javadikooshesh, S.; Zarrinrad, G.; Esmaeili, F.; Alishahi, Z.; Sabourinejad, Z.; et al. IL-6/IL-6R pathway is a therapeutic target in chemoresistant ovarian cancer. Tumori 2019, 105, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Nemeth, J.; O’Brien, C.; Susa, M.; Liu, X.; Zhang, Z.; Choy, E.; Mankin, H.; Hornicek, F.; Duan, Z. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin. Cancer Res. 2010, 16, 5759–5769. [Google Scholar] [CrossRef]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Wouters, M.C.; Kenter, G.G.; van Erkel, A.R.; van Poelgeest, M.I.; Nijman, H.W.; van der Hoeven, J.J.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef]
- Andersen, C.L.; Sikora, M.J.; Boisen, M.M.; Ma, T.; Christie, A.; Tseng, G.; Park, Y.; Luthra, S.; Chandran, U.; Haluska, P.; et al. Active Estrogen Receptor-alpha Signaling in Ovarian Cancer Models and Clinical Specimens. Clin. Cancer Res. 2017, 23, 3802–3812. [Google Scholar] [CrossRef]
- Kreuzinger, C.; von der Decken, I.; Wolf, A.; Gamperl, M.; Koller, J.; Karacs, J.; Pfaffinger, S.; Bartl, T.; Reinthaller, A.; Grimm, C.; et al. Patient-derived cell line models revealed therapeutic targets and molecular mechanisms underlying disease progression of high grade serous ovarian cancer. Cancer Lett. 2019, 459, 1–12. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, T. Biological characterization including sensitivity to mitomycin C of cultured human ovarian cancers. Nippon Sanka Fujinka Gakkai Zasshi 1981, 33, 1197–1204. [Google Scholar] [PubMed]
- Yanagibashi, T.; Gorai, I.; Nakazawa, T.; Miyagi, E.; Hirahara, F.; Kitamura, H.; Minaguchi, H. Complexity of expression of the intermediate filaments of six new human ovarian carcinoma cell lines: New expression of cytokeratin 20. Br. J. Cancer 1997, 76, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator V3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Hall, M.D.; Telma, K.A.; Chang, K.E.; Lee, T.D.; Madigan, J.P.; Lloyd, J.R.; Goldlust, I.S.; Hoeschele, J.D.; Gottesman, M.M. Say no to DMSO: Dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res. 2014, 74, 3913–3922. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Km [nM] | Vmax [fmol/106 Cells/h] | ||||
|---|---|---|---|---|---|---|
| DHEA-S | AD | T | DHEA-S | AD | T | |
| Carboplatin-sensitive | ||||||
| 13363 | 193.9 ± 9.3 | 812.7 ± 79.7 | 657.7 ± 52.4 | 3523.7 ± 47.8 | 1736.6 ± 75.9 | 85.6 ± 2.8 |
| 13699 | 308.7 ± 19.2 | 929.4 ± 68.8 | 623.5 ± 50.2 | 3192.4 ± 64.7 | 1478.8 ± 51.1 | 51.4 ± 1.7 |
| Carboplatin-resistant | ||||||
| 13914_1 | 318.7 ± 18.6 | 854.1 ± 39.9 | n.c. | 729.0 ± 13.9 * | 269.8 ± 5.7 * | 0.0 * |
| 15233 | 314.1 ± 11.1 | 980.4 ± 88.6 | 592.0 ± 33.0 | 328.5 ± 3.8 * | 369.7 ± 15.9 * | 12.3 ± 0.3 * |
| Kuramochi | 318.5 ± 36.2 | 841.7 ± 43.4 | 621.5 ± 45.4 | 56.9 ± 2.1 * | 189.0 ± 4.4* | 3.4 ± 0.1 * |
| OVSAHO | 310.8 ± 20.7 | 871.4 ± 36.7 | 631.4 ± 30.8 | 264.8 ± 5.7 * | 204.9 ± 3.9 * | 4.4 ± 0.1 * |
| Km [nM] | ||||
|---|---|---|---|---|
| Cell line | E2 | E1-S | E2-S | E2-G |
| Carboplatin-sensitive | ||||
| 13363 | 430.4 ± 22.9 | 390.5 ± 18.4 | 426.8 ± 22.1 | 580.1 ± 20.0 |
| 13699 | 434.8 ± 27.9 | 399.3 ± 25.5 | 422.5 ± 29.8 | 532.9 ± 34.3 |
| Carboplatin-resistant | ||||
| 13914_1 | 440.8 ± 13.2 | 398.0 ± 9.1 | 418.0 ± 16.8 | 535.3 ± 39.6 |
| 15233 | 451.4 ± 22.8 | 407.3 ± 29.0 | 458.8 ± 38.7 | 537.0 ± 44.3 |
| Kuramochi | 454.0 ± 23.4 | 402.8 ± 14.1 | 452.0 ± 23.7 | 521.2 ± 52.6 |
| OVSAHO | 450.0 ± 19.8 | 403.3 ±18.3 | 464.6 ± 54.5 | 534.4 ± 48.5 |
| Vmax (fmol/106 Cells/h) | ||||
| Cell line | E2 | E1-S | E2-S | E2-G |
| Carboplatin-sensitive | ||||
| 13363 | 6492.9 ± 124.7 | 2404.8 ± 39.7 | 198.3 ± 3.7 | 30.0 ± 0.4 |
| 13699 | 6028.7 ± 140.4 | 2067.9 ± 46.6 | 373.7 ± 9.5 | 24.3 ± 0.6 |
| Carboplatin-resistant | ||||
| 13914_1 | 4687.8 ± 51.2 * | 318.9 ± 2.6 * | 21.5 ± 0.3 * | 3.3 ± 0.1 * |
| 15233 | 4017.7 ± 74.5 * | 455.7 ± 11.5 * | 38.8 ± 1.2 * | 2.8 ± 0.1 * |
| Kuramochi | 3750.0 ± 71.0 * | 508.4 ± 6.3 * | 42.7 ± 0.8 * | 3.5 ± 0.1 * |
| OVSAHO | 3833.8 ± 61.9 * | 443.9 ± 7.1 * | 70.8 ± 3.1 * | 3.9 ± 0.1 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poschner, S.; Wackerlig, J.; Castillo-Tong, D.C.; Wolf, A.; von der Decken, I.; Rižner, T.L.; Pavlič, R.; Meshcheryakova, A.; Mechtcheriakova, D.; Fritzer-Szekeres, M.; et al. Metabolism of Estrogens: Turnover Differs between Platinum-Sensitive and -Resistant High-Grade Serous Ovarian Cancer Cells. Cancers 2020, 12, 279. https://doi.org/10.3390/cancers12020279
Poschner S, Wackerlig J, Castillo-Tong DC, Wolf A, von der Decken I, Rižner TL, Pavlič R, Meshcheryakova A, Mechtcheriakova D, Fritzer-Szekeres M, et al. Metabolism of Estrogens: Turnover Differs between Platinum-Sensitive and -Resistant High-Grade Serous Ovarian Cancer Cells. Cancers. 2020; 12(2):279. https://doi.org/10.3390/cancers12020279
Chicago/Turabian StylePoschner, Stefan, Judith Wackerlig, Dan Cacsire Castillo-Tong, Andrea Wolf, Isabel von der Decken, Tea Lanišnik Rižner, Renata Pavlič, Anastasia Meshcheryakova, Diana Mechtcheriakova, Monika Fritzer-Szekeres, and et al. 2020. "Metabolism of Estrogens: Turnover Differs between Platinum-Sensitive and -Resistant High-Grade Serous Ovarian Cancer Cells" Cancers 12, no. 2: 279. https://doi.org/10.3390/cancers12020279
APA StylePoschner, S., Wackerlig, J., Castillo-Tong, D. C., Wolf, A., von der Decken, I., Rižner, T. L., Pavlič, R., Meshcheryakova, A., Mechtcheriakova, D., Fritzer-Szekeres, M., Thalhammer, T., & Jäger, W. (2020). Metabolism of Estrogens: Turnover Differs between Platinum-Sensitive and -Resistant High-Grade Serous Ovarian Cancer Cells. Cancers, 12(2), 279. https://doi.org/10.3390/cancers12020279