Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma


 ,
, 

Abstract
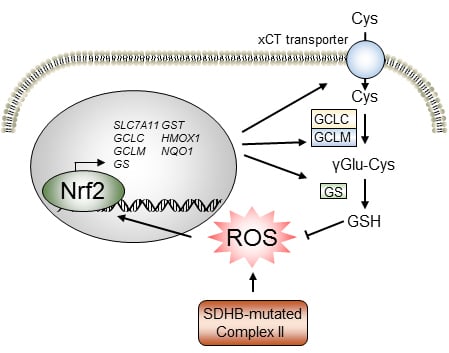
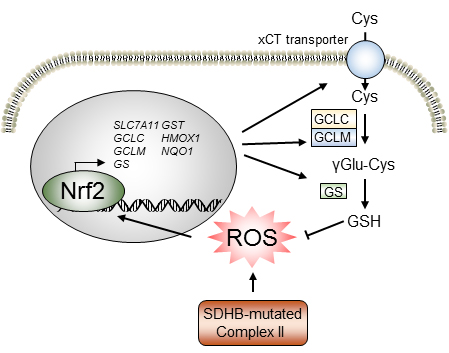{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
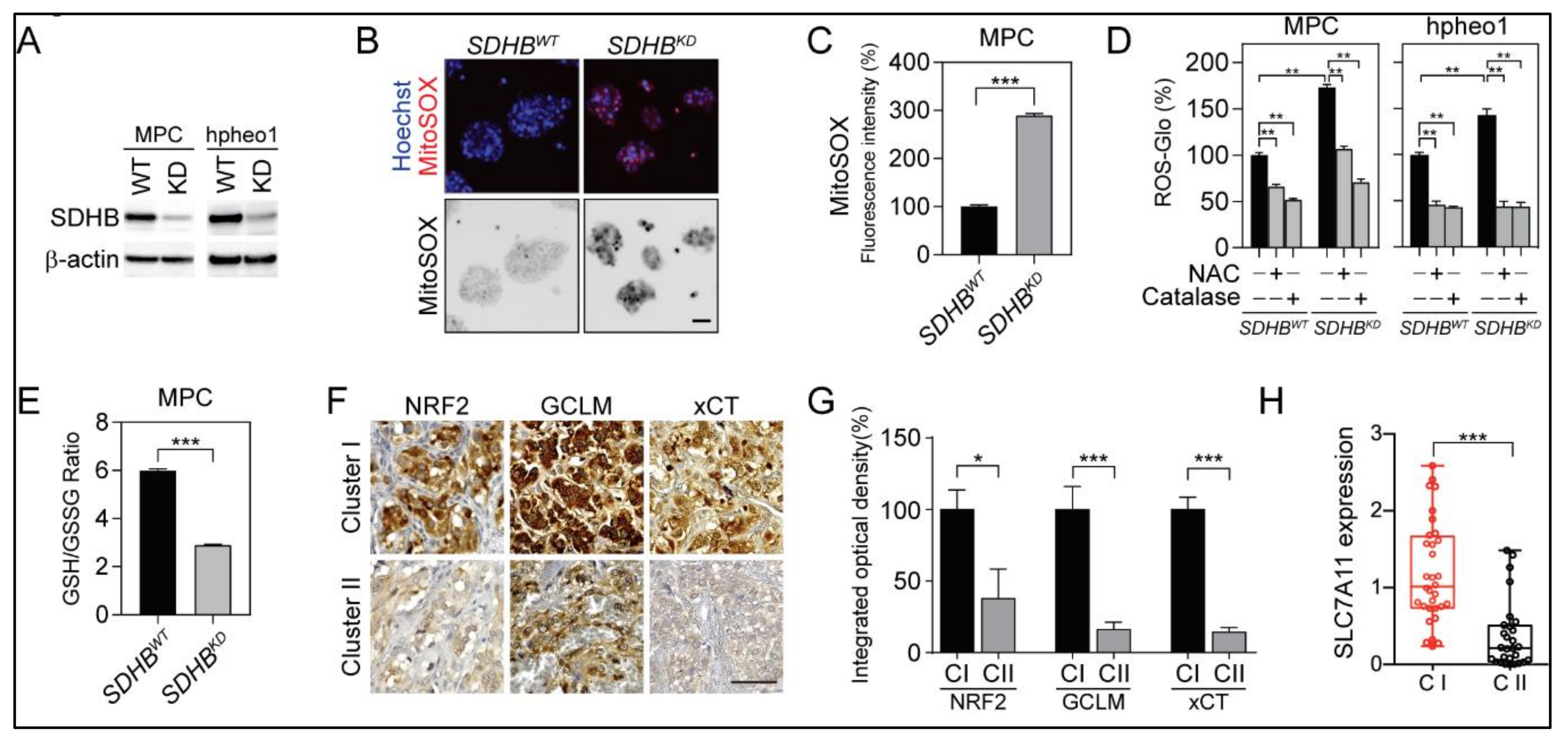
2.1. SDHB Deficiency Altered the Redox Balance in PCPG Cells
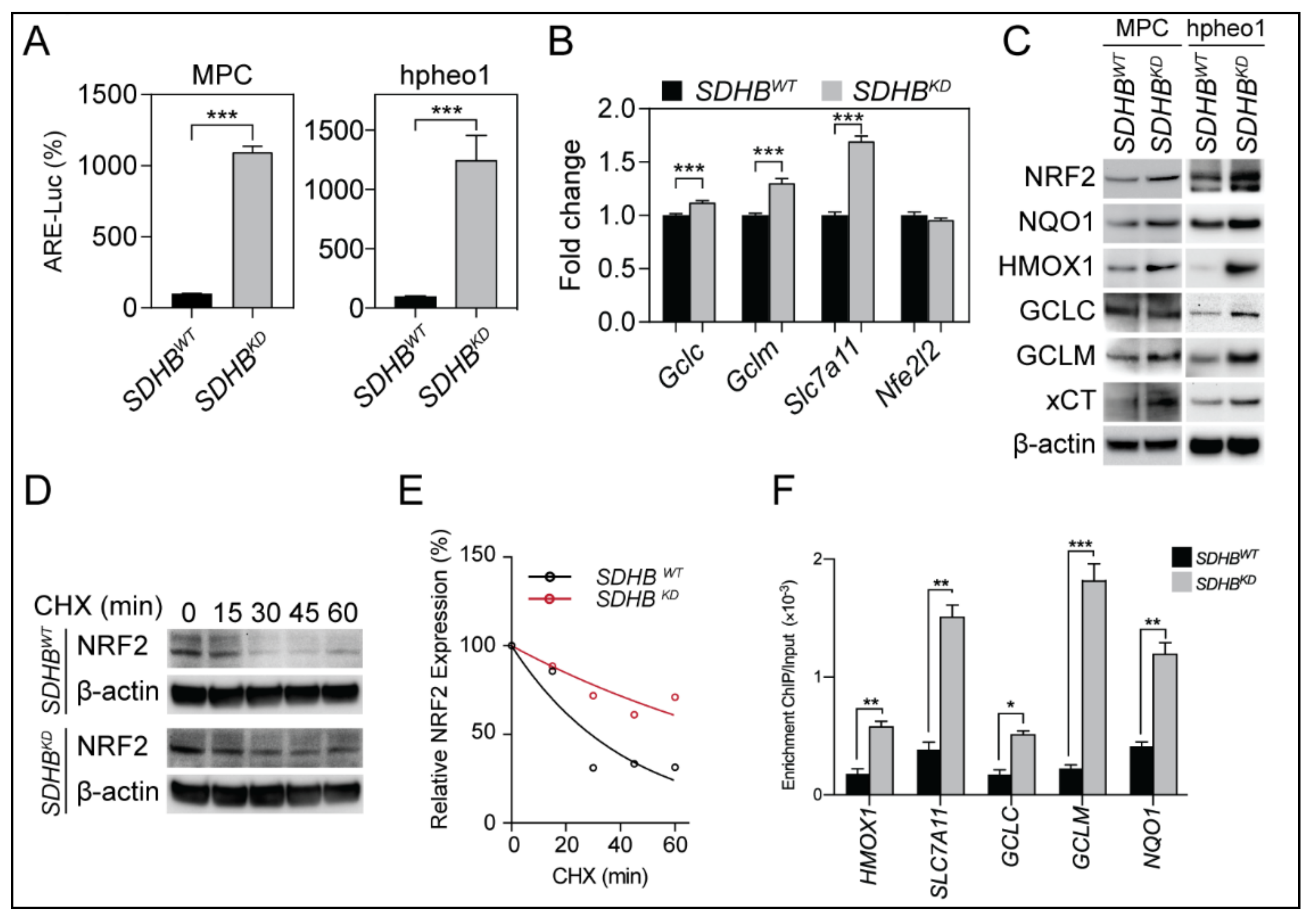
2.2. NRF2 Supported Glutathione De Novo Synthesis in SDHBKD Cells
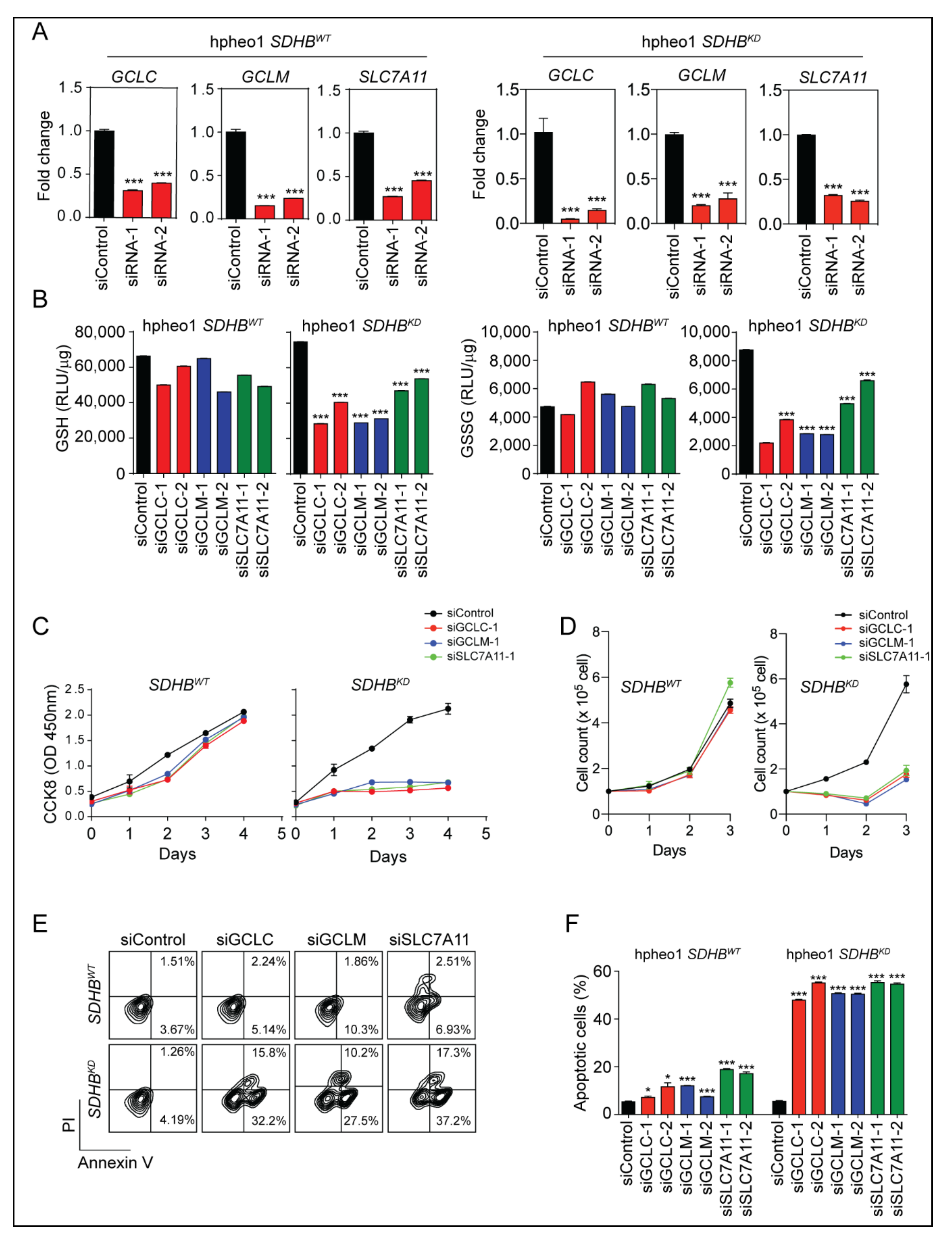
2.3. Glutathione Synthesis Protected SDHBKD Cells
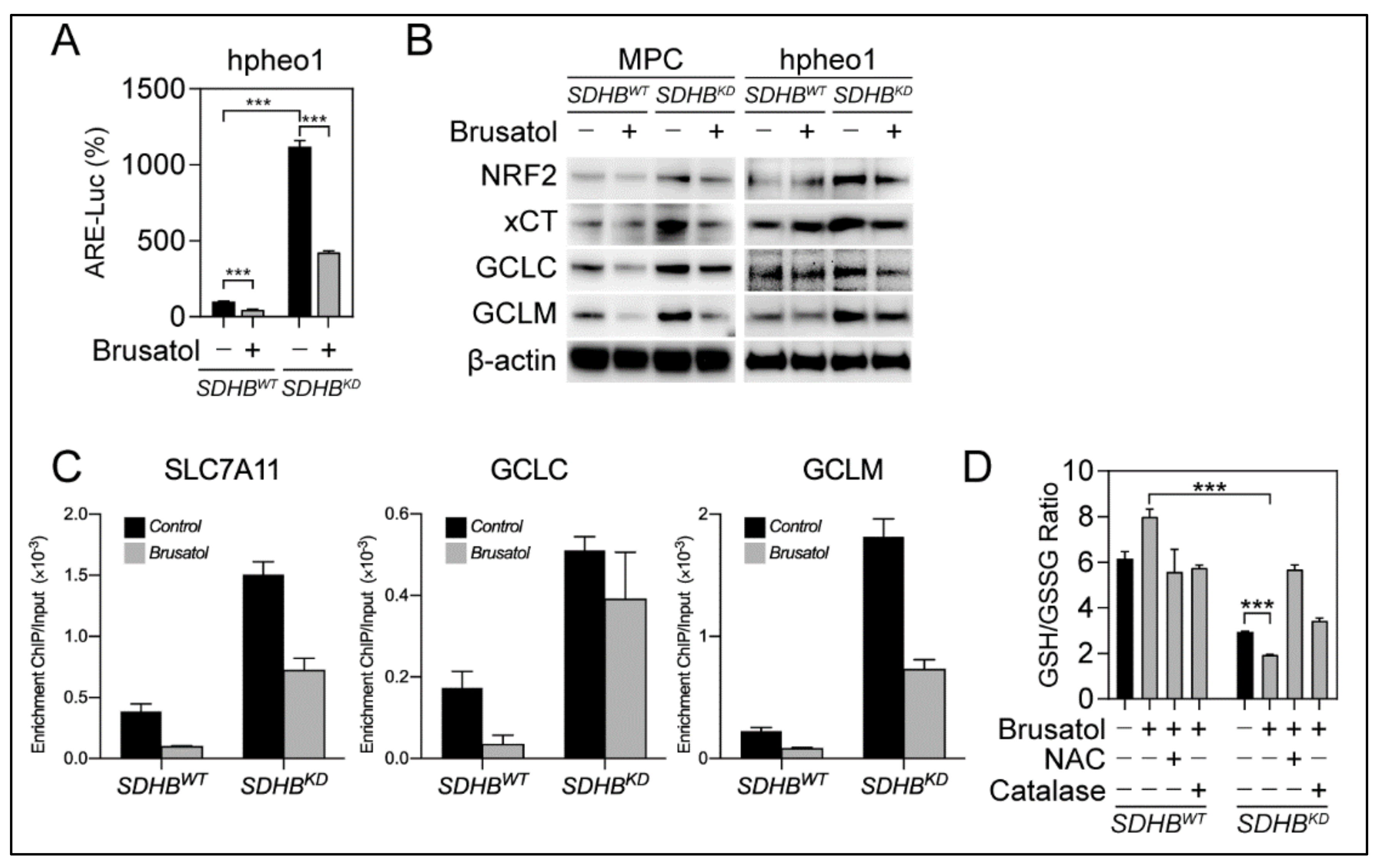
2.4. The NRF2 Inhibitor Brusatol Disrupted Glutathione Synthesis
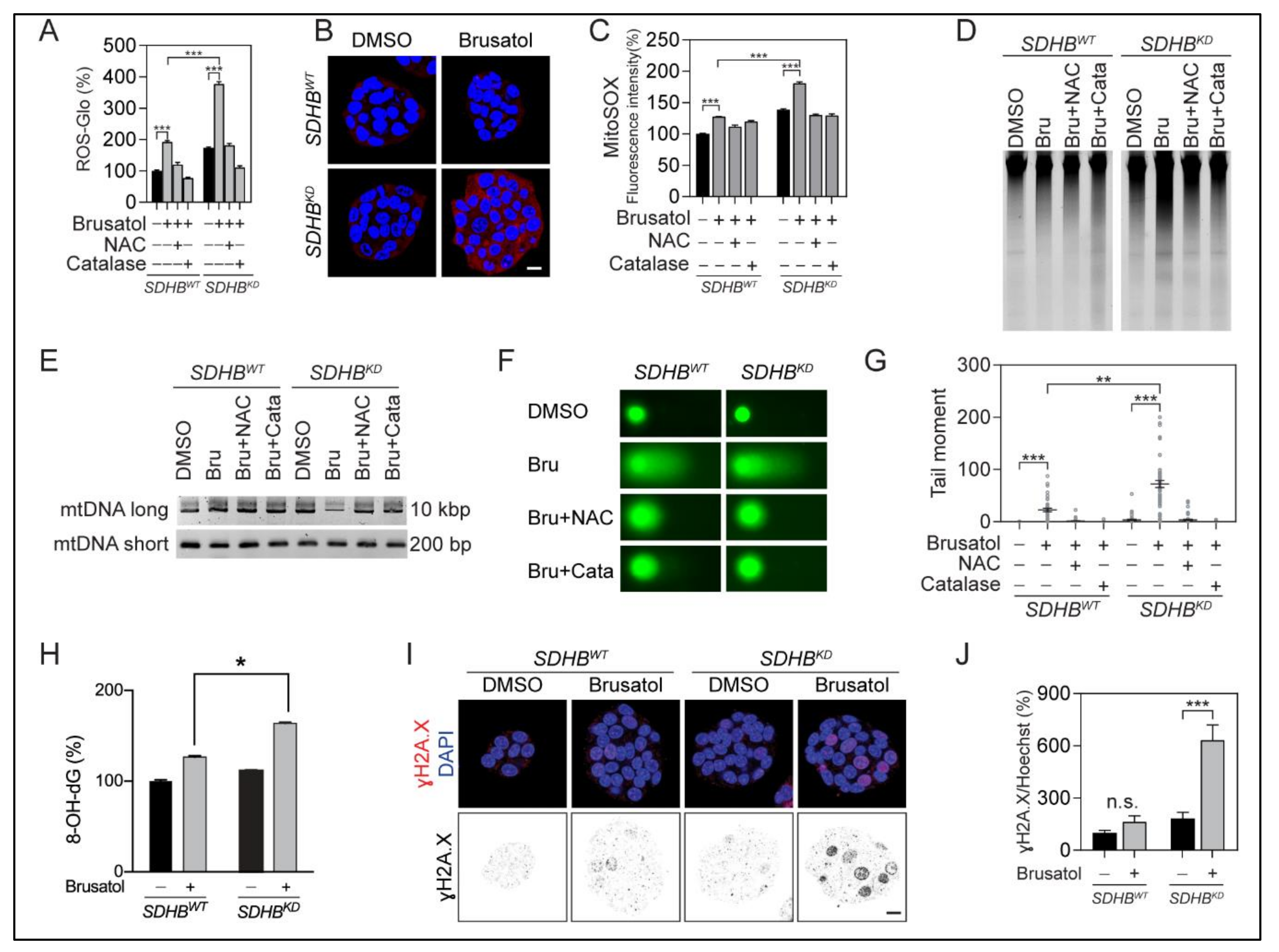
2.5. NRF2 Suppression Led to Oxidation-Derived Cellular Damage
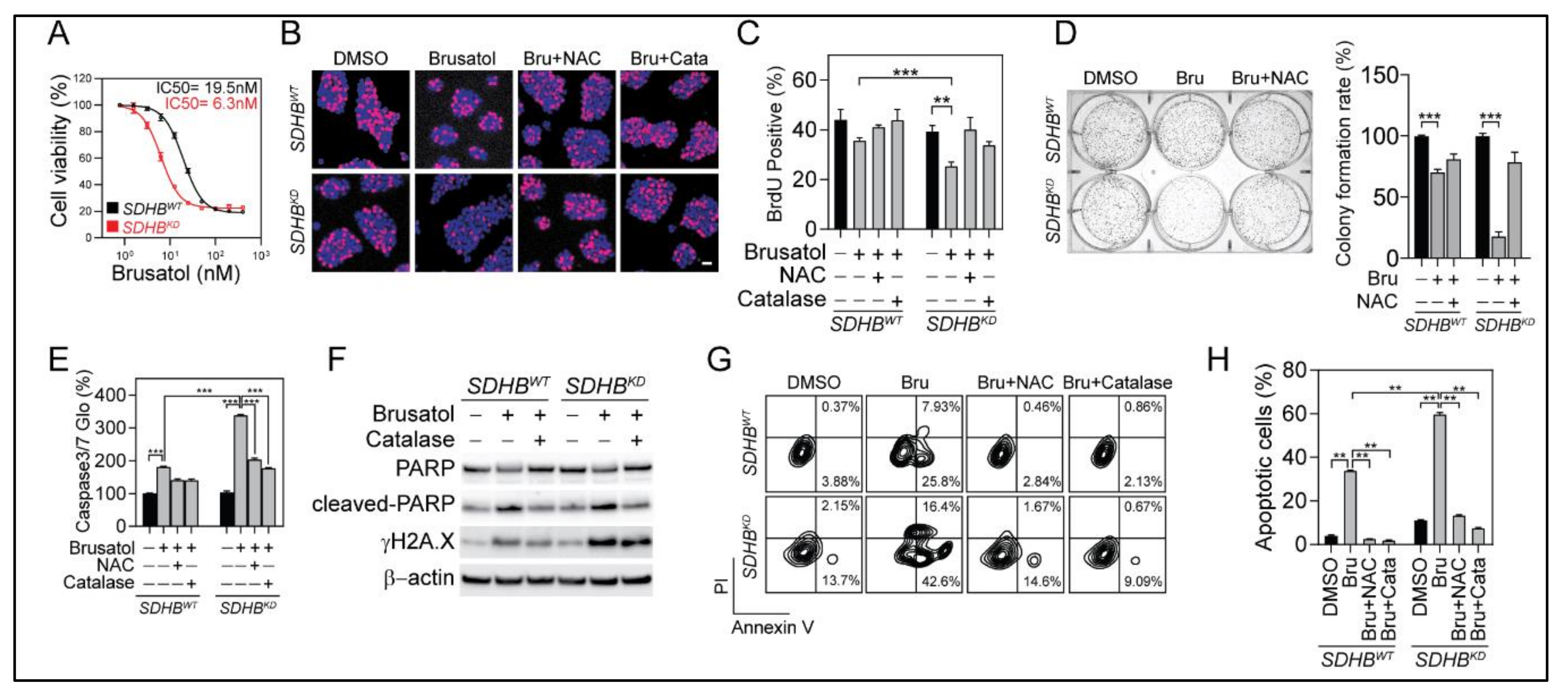
2.6. Selective Vulnerability of SDHBKD Cells to NRF2 Suppression
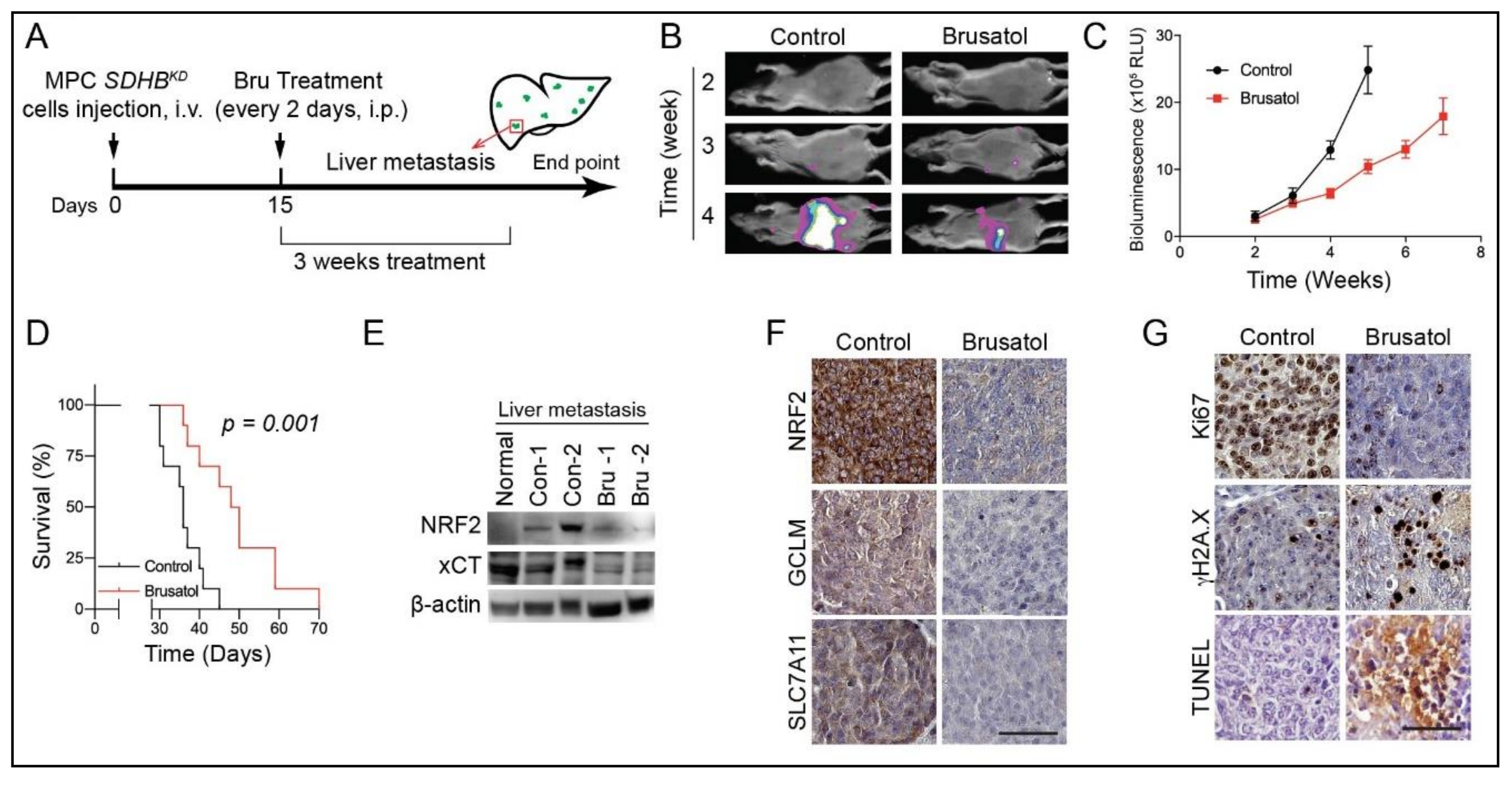
2.7. NRF2 Blockade for SDHB-Mutated PCPGs
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Patient Samples
4.3. Cell Lines
4.4. Reagents and Treatment Condition
4.5. Real-Time PCR
4.6. RNA Interference
4.7. Chromatin Immunoprecipitation (ChIP) Assay
4.8. ROS Quantification
4.9. ARE Luciferase Reporter Assay
4.10. Immunoblotting
4.11. Immunofluorescence Staining
4.12. Annexin V/PI Apoptosis Assay
4.13. Caspase-3/7 Activity Assay
4.14. DNA Oxidative Damage ELISA Assay
4.15. DNA Fragmentation Assay
4.16. Comet Assay
4.17. BrdU-Incorporated Cell Proliferation
4.18. Colony Formation Assay
4.19. Cell Viability Analysis
4.20. Immunoprecipitation and Ubiquitination Assay
4.21. CHX Pulse Chase Protein Half-Life Measurement
4.22. Metastatic Allograft Mouse Model
4.23. Immunohistochemistry and TUNEL Assay
4.24. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castro-Vega, L.J.; Letouze, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Liu, Y.; Pacak, K.; Yang, C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers 2019, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Borozdin, W.; Mautner, V.F.; Hoffmann, M.M.; Boehm, D.; Robledo, M.; Cascon, A.; Harenberg, T.; Schiavi, F.; Pawlu, C.; et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2007, 92, 2784–2792. [Google Scholar] [CrossRef]
- Jochmanova, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Fishbein, L.; Khare, S.; Wubbenhorst, B.; DeSloover, D.; D’Andrea, K.; Merrill, S.; Cho, N.W.; Greenberg, R.A.; Else, T.; Montone, K.; et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6140. [Google Scholar] [CrossRef]
- Job, S.; Draskovic, I.; Burnichon, N.; Buffet, A.; Cros, J.; Lepine, C.; Venisse, A.; Robidel, E.; Verkarre, V.; Meatchi, T.; et al. Telomerase activation and ATRX mutations are independent risk factors for metastatic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2018, 25, 760–770. [Google Scholar] [CrossRef]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef] [PubMed]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.A.; et al. Characteristics and Outcomes of Metastatic Sdhb and Sporadic Pheochromocytoma/Paraganglioma: An National Institutes of Health Study. Endocr. Pract. 2016, 22, 302–314. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Nomura, K.; Kimura, H.; Shimizu, S.; Kodama, H.; Okamoto, T.; Obara, T.; Takano, K. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J. Clin. Endocrinol. Metab. 2009, 94, 2850–2856. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Alblas, G.; van Hulsteijn, L.T.; Dekkers, O.M.; Corssmit, E.P. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 81, 642–651. [Google Scholar] [CrossRef]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD+/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef]
- Owens, K.M.; Aykin-Burns, N.; Dayal, D.; Coleman, M.C.; Domann, F.E.; Spitz, D.R. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2−• and H2O2. Free Radic. Biol. Med. 2012, 52, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Yankovskaya, V.; Horsefield, R.; Tornroth, S.; Luna-Chavez, C.; Miyoshi, H.; Leger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Wen, H.; Dong, L.; Yan, B.; Vider, J.; Boukalova, S.; Krobova, L.; Vanova, K.; Zobalova, R.; Sobol, M.; et al. Alternative assembly of respiratory complex II connects energy stress to metabolic checkpoints. Nat. Commun. 2018, 9, 2221. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Fu, X.; Liu, Y.; Yu, D.; Cai, S.J.; Yang, C. Blockade of Glutathione Metabolism in IDH1-Mutated Glioma. Mol. Cancer Ther. 2019, 19, 221–230. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Liu, Q. Brusatol Enhances the Radiosensitivity of A549 Cells by Promoting ROS Production and Enhancing DNA Damage. Int. J. Mol. Sci. 2016, 17, 997. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Lai, Z.Q.; Leung, A.W.N.; Leung, P.S.; Li, Z.S.; Lin, Z.X. Exploring brusatol as a new anti-pancreatic cancer adjuvant: Biological evaluation and mechanistic studies. Oncotarget 2017, 8, 84974–84985. [Google Scholar] [CrossRef] [PubMed]
- Ghayee, H.K.; Bhagwandin, V.J.; Stastny, V.; Click, A.; Ding, L.H.; Mizrachi, D.; Zou, Y.S.; Chari, R.; Lam, W.L.; Bachoo, R.M.; et al. Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS ONE 2013, 8, e65624. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Fujita, K.; Tagawa, K.; Chen, X.; Homma, H.; Sasabe, T.; Shimizu, J.; Shimizu, S.; Tamura, T.; Muramatsu, S.; et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 2015, 7, 78–101. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Yang, C. Evaluating In Vitro DNA Damage Using Comet Assay. J. Vis. Exp. 2017, 128, e56450. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sun, M.G.; Matro, J.; Huynh, T.T.; Rahimpour, S.; Prchal, J.T.; Lechan, R.; Lonser, R.; Pacak, K.; Zhuang, Z. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood 2013, 121, 2563–2566. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Zhu, D.; Hong, C.S.; Dmitriev, P.; Zhang, C.; Li, Y.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation. Proc. Natl. Acad. Sci. USA 2015, 112, 1137–1142. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Pang, Y.; Caisova, V.; Ding, J.; Yu, D.; Zhou, Y.; Huynh, T.-T.; Ghayee, H.; Pacak, K.; Yang, C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers 2020, 12, 280. https://doi.org/10.3390/cancers12020280
Liu Y, Pang Y, Caisova V, Ding J, Yu D, Zhou Y, Huynh T-T, Ghayee H, Pacak K, Yang C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers. 2020; 12(2):280. https://doi.org/10.3390/cancers12020280
Chicago/Turabian StyleLiu, Yang, Ying Pang, Veronika Caisova, Jianyi Ding, Di Yu, Yiqiang Zhou, Thanh-Truc Huynh, Hans Ghayee, Karel Pacak, and Chunzhang Yang. 2020. "Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma" Cancers 12, no. 2: 280. https://doi.org/10.3390/cancers12020280
APA StyleLiu, Y., Pang, Y., Caisova, V., Ding, J., Yu, D., Zhou, Y., Huynh, T.-T., Ghayee, H., Pacak, K., & Yang, C. (2020). Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers, 12(2), 280. https://doi.org/10.3390/cancers12020280