Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance

 , ,
, , 

Abstract
Simple Summary
Abstract
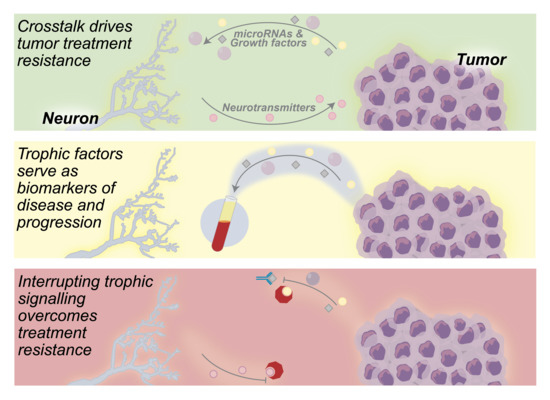
1. Introduction
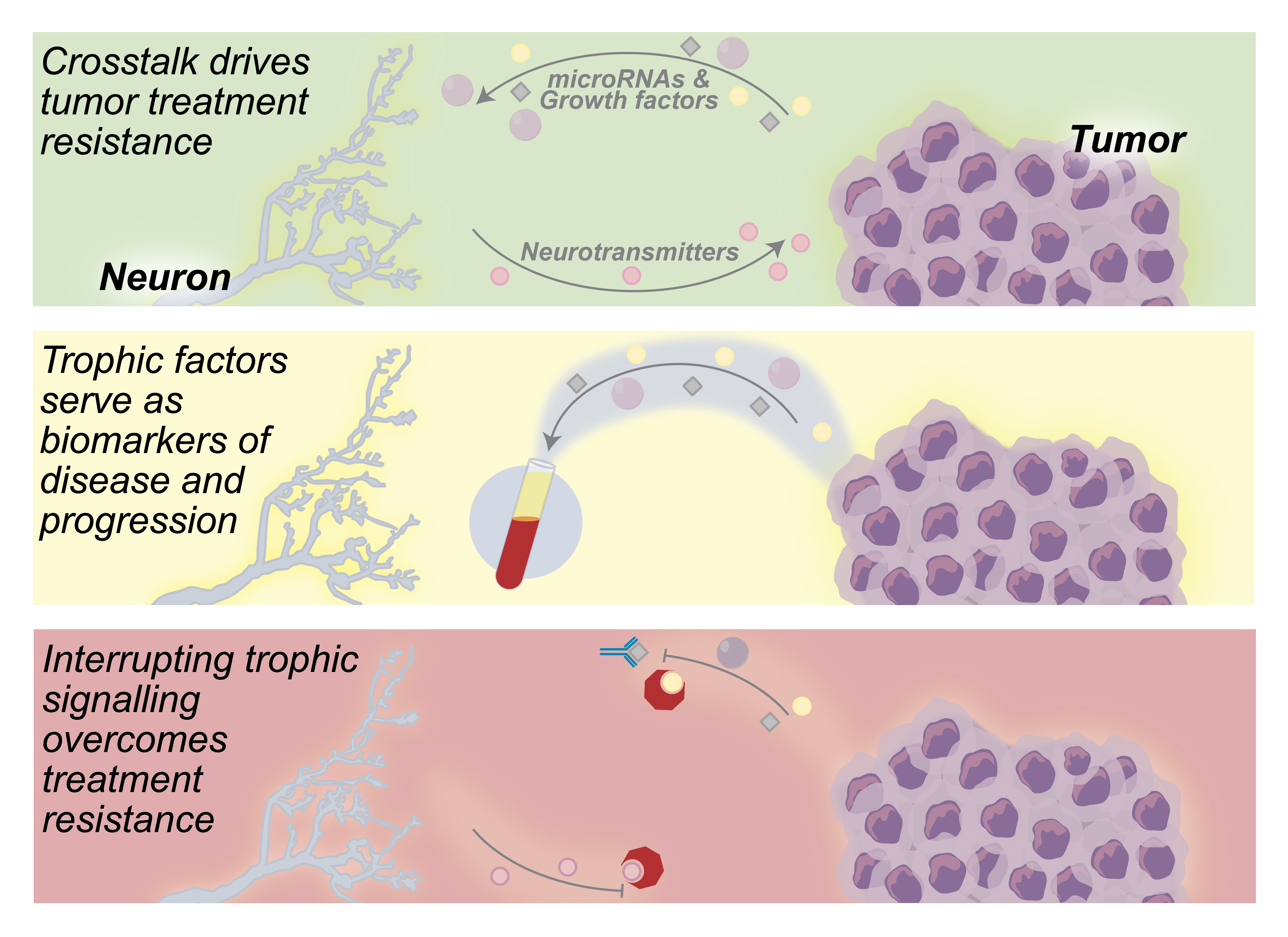
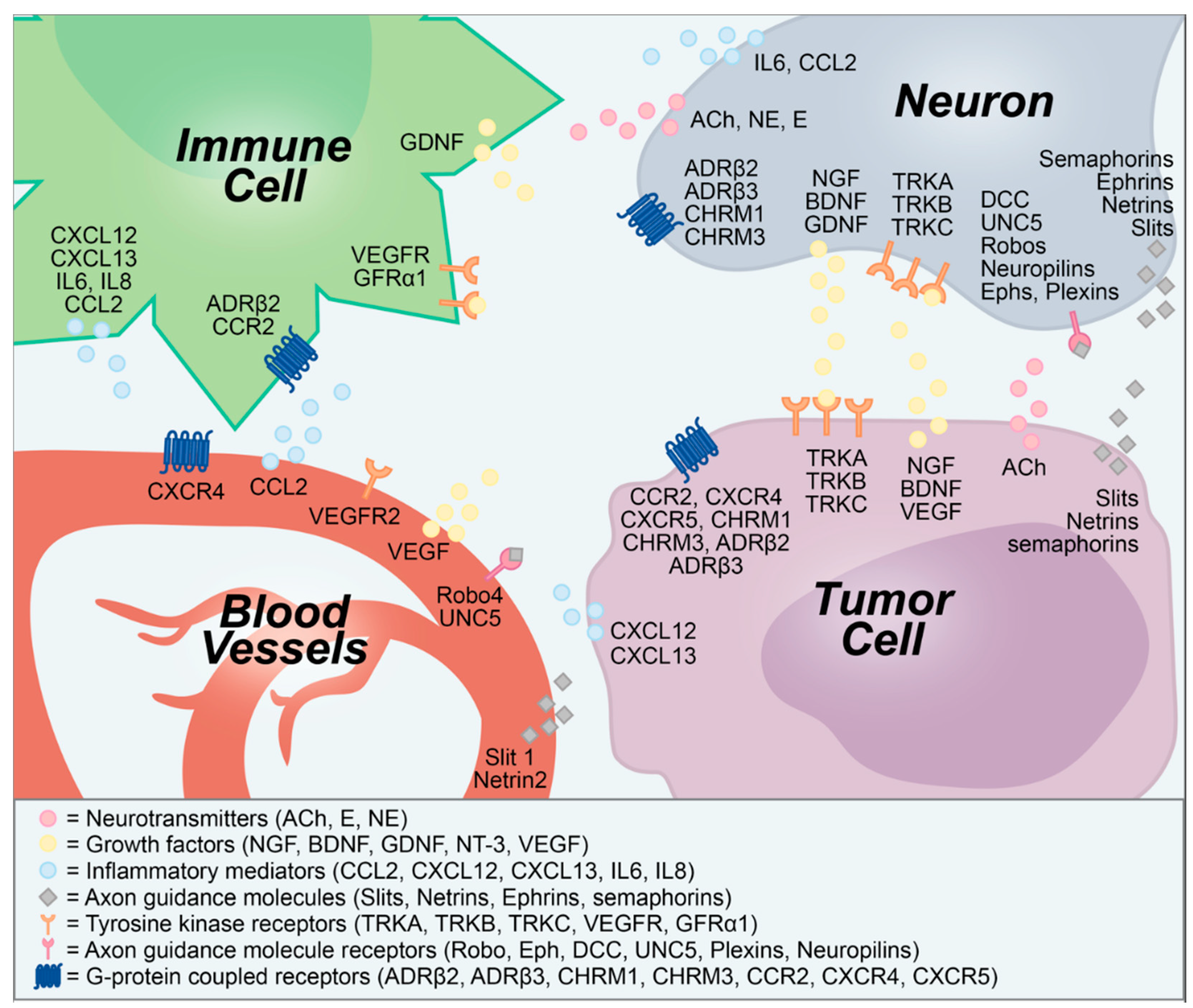
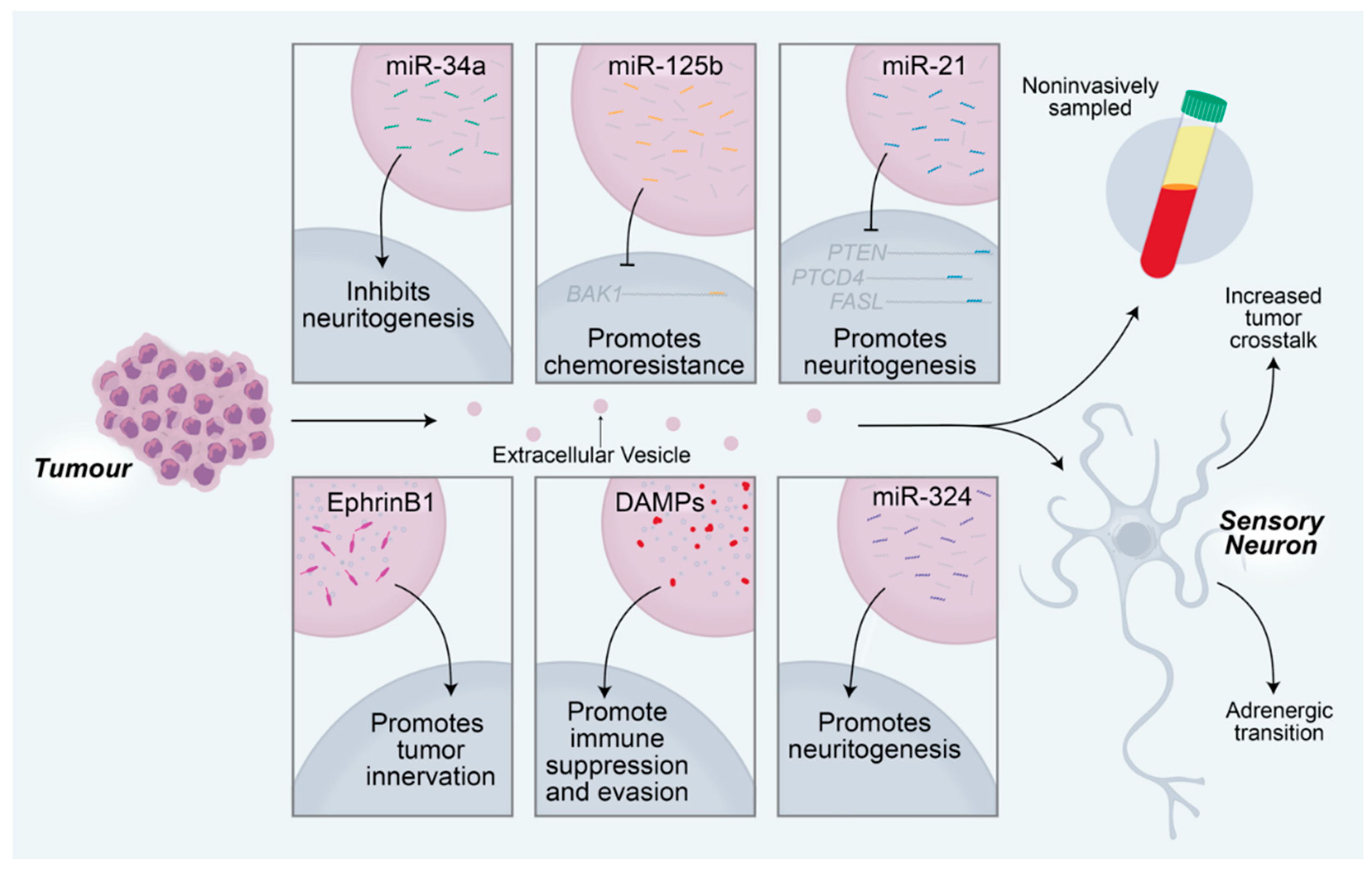
2. Neuron—Tumor Communication Drives Tumor Growth
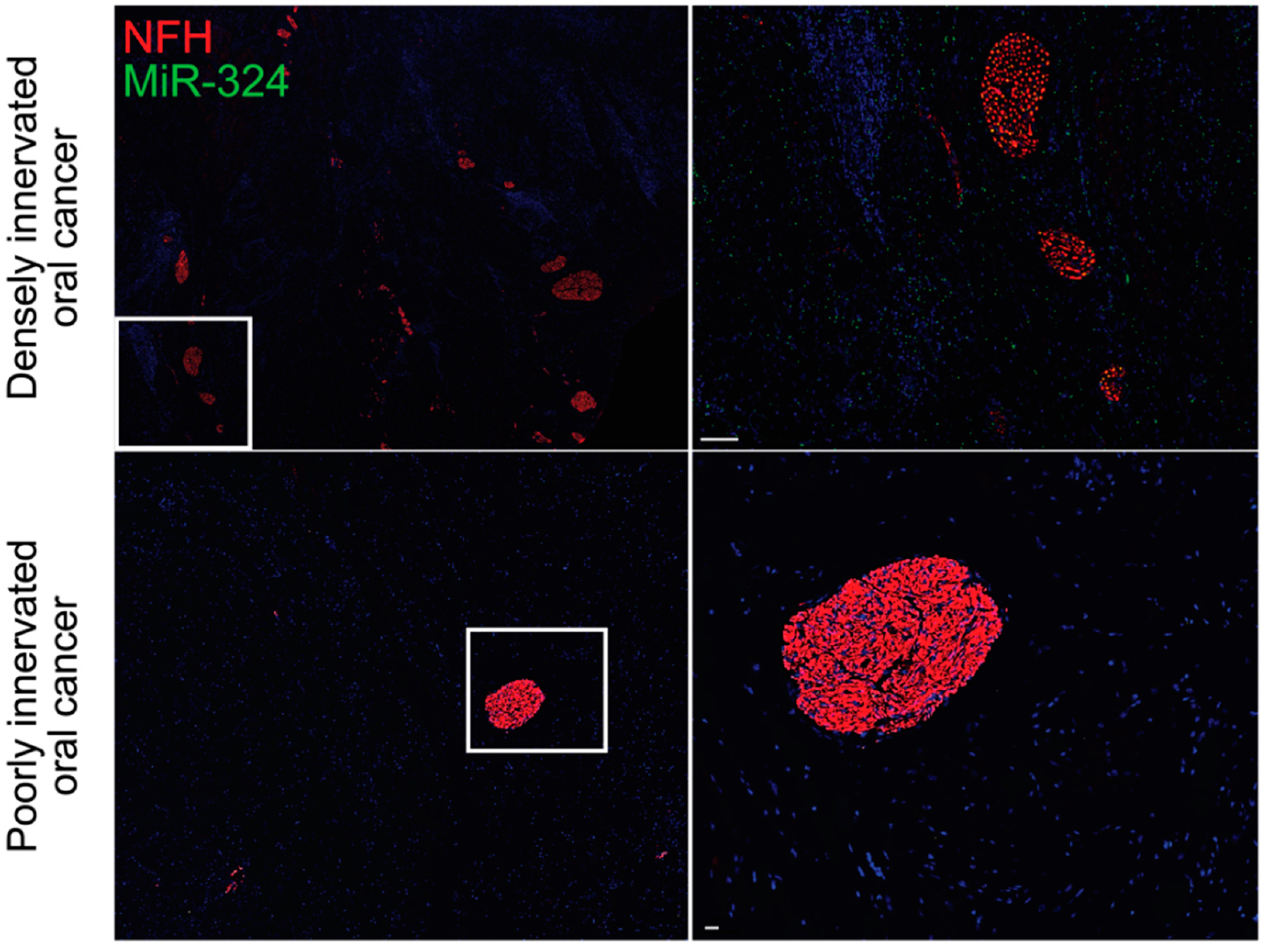
3. Neurogenic and Oncogenic Trophic Factors as Biomarkers of Disease
4. Neuron—Tumor Signaling as a Target for Anticancer Therapeutics
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbink, M.R.; van Deijk, A.L.F.; Heine, V.M.; Verheijen, M.H.; Korosi, A. The involvement of astrocytes in early-life adversity induced programming of the brain. Glia 2019, 67, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.A.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Hartenstein, V.; Stollewerk, A. The evolution of early neurogenesis. Dev. Cell 2015, 32, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Park, H.T.; Wu, J.; Rao, Y. Molecular control of neuronal migration. BioEssays 2002, 24, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Bixby, J.L.; Harris, W.A. Molecular mechanisms of axon growth and guidance. Annu. Rev. Cell Biol. 1991, 7, 117–159. [Google Scholar] [CrossRef]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Gould, E.; Reeves, A.J.; Graziano, M.S.A.; Gross, C.G. Neurogenesis in the neocortex of adult primates. Science 1999, 286, 548–552. [Google Scholar] [CrossRef]
- Bernier, P.J.; Bédard, A.; Vinet, J.; Lévesque, M.; Parent, A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc. Natl. Acad. Sci. USA 2002, 99, 11464–11469. [Google Scholar] [CrossRef]
- Zhao, M.; Momma, S.; Delfani, K.; Carlén, M.; Cassidy, R.M.; Johansson, C.B.; Brismar, H.; Shupliakov, O.; Frisén, J.; Janson, A.M. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. USA 2003, 100, 7925–7930. [Google Scholar] [CrossRef]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G.; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Kater, S.B. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989, 12, 265–270. [Google Scholar] [CrossRef]
- Henderson, C.E. Role of neurotrophic factors in neuronal development. Curr. Opin. Neurobiol. 1996, 6, 64–70. [Google Scholar] [CrossRef]
- Quaegebeur, A.; Lange, C.; Carmeliet, P. The neurovascular link in health and disease: Molecular mechanisms and therapeutic implications. Neuron 2011, 71, 406–424. [Google Scholar] [CrossRef]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar] [CrossRef]
- Segarra, M.; Aburto, M.R.; Hefendehl, J.; Acker-Palmer, A. Neurovascular interactions in the nervous system. Annu. Rev. Cell Dev. Biol. 2019, 35, 615–635. [Google Scholar] [CrossRef]
- Kumar, A.; Brockes, J.P. Nerve dependence in tissue, organ, and appendage regeneration. Trends Neurosci. 2012, 35, 691–699. [Google Scholar] [CrossRef]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta Rev. Cancer 2011, 1816, 105–118. [Google Scholar] [CrossRef]
- Venkatesh, H.; Monje, M. Neuronal activity in ontogeny and oncology. Trends Cancer 2017, 3, 89–112. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Liebig, C.; Ayala, G.; Wilks, J.A.; Berger, D.H.; Albo, D. Perineural invasion in cancer: A review of the literature. Cancer 2009, 115, 3379–3391. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Na’Ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Rodin, A.E.; Larson, D.L.; Roberts, D.K. Nature of the perineural space invaded by prostatic carcinoma. Cancer 1967, 20, 1772–1779. [Google Scholar] [CrossRef]
- Hassan, M.O.; Maksem, J. The prostatic perineural space and its relation to tumor spread. Am. J. Surg. Pathol. 1980, 4, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Bakst, R.L.; Wong, R.J. Mechanisms of Perineural Invasion. J. Neurol. Surg. Part B Skull Base 2016, 77, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Li, L.; Hanahan, D. Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 2013, 153, 86–100. [Google Scholar] [CrossRef]
- Li, L.; Zeng, Q.; Bhutkar, A.; Galván, J.A.; Karamitopoulou, E.; Noordermeer, D.; Peng, M.W.; Piersigilli, A.; Perren, A.; Zlobec, I.; et al. GKAP Acts as a Genetic Modulator of NMDAR Signaling to Govern Invasive Tumor Growth. Cancer Cell 2018, 33, 736–751.e5. [Google Scholar] [CrossRef]
- Robinson, H.P.C.; Li, L. Autocrine, paracrine and necrotic NMDA receptor signalling in mouse pancreatic neuroendocrine tumour cells. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Ayala, G.E.; Wheeler, T.M.; David Shine, H.; Schmelz, M.; Frolov, A.; Chakraborty, S.; Rowley, D. In vitro dorsal root ganglia and human prostate cell line interaction: Redefining perineural invasion in prostate cancer. Prostate 2001, 49, 213–223. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; McAlhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res. 2002, 62, 3298–3307. [Google Scholar] [PubMed]
- Cavel, O.; Shomron, O.; Shabtay, A.; Vital, J.; Trejo-Leider, L.; Weizman, N.; Krelin, Y.; Fong, Y.; Wong, R.J.; Amit, M.; et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. 2012, 72, 5733–5743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qi, L.; Li, M.; Zhang, D.; Xu, S.; Wang, N.; Sun, B. Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J. Exp. Clin. Cancer Res. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, X.; Guo, H.; Fu, L.; Pan, G.; Sun, Y. CXCL13-CXCR5 axis promotes the growth and invasion of colon cancer cells via PI3K/AKT pathway. Mol. Cell. Biochem. 2015, 400, 287–295. [Google Scholar] [CrossRef]
- He, S.; He, S.; Chen, C.H.; Deborde, S.; Bakst, R.L.; Chernichenko, N.; McNamara, W.F.; Lee, S.Y.; Barajas, F.; Yu, Z.; et al. The chemokine (CCL2-CCR2) signaling axis mediates perineural invasion. Mol. Cancer Res. 2015, 13, 380–390. [Google Scholar] [CrossRef]
- Voss, M.J.; Entschladen, F. Tumor interactions with soluble factors and the nervous system. Cell Commun. Signal. 2010, 8, 1–6. [Google Scholar] [CrossRef]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain. Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Zue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 12363611–12363619. [Google Scholar] [CrossRef]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; Li, Y.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress hormone-mediated invasion of ovarian cancer cells. Clin. Cancer Res. 2006, 12, 369–375. [Google Scholar] [CrossRef]
- Drell IV, T.L.; Joseph, J.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res. Treat. 2003, 80, 63–70. [Google Scholar] [CrossRef]
- Masur, K.; Niggemann, B.; Zanker, K.S.; Entschladen, F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by β-blockers. Cancer Res. 2001, 61, 2866–2869. [Google Scholar] [PubMed]
- Guo, K.; Ma, Q.; Li, J.; Wang, Z.; Shan, T.; Li, W.; Xu, Q.; Xie, K. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of STAT3 signaling. Mol. Cancer Ther. 2013, 12, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Chakroborty, D.; Basu, S. Neurotransmitters as Regulators of Tumor Angiogenesis and Immunity: The Role of Catecholamines. J. Neuroimmune Pharmacol. 2013, 8, 7–14. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Armaiz-Pena, G.; Takahashi, R.; Lin, Y.G.; Trevino, J.; Li, Y.; Jennings, N.; Arevalo, J.; Lutgendorf, S.K.; Gallick, G.E.; et al. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J. Biol. Chem. 2007, 282, 29919–29926. [Google Scholar] [CrossRef]
- Shahzad, M.M.K.; Arevalo, J.M.; Armaiz-Pena, G.N.; Lu, C.; Stone, R.L.; Moreno-Smith, M.; Nishimura, M.; Lee, J.W.; Jennings, N.B.; Bottsford-Miller, J.; et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. J. Biol. Chem. 2010, 285, 35462–35470. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.G.; Morizono, K.; Karanikolas, B.D.W.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef]
- Inbar, S.; Neeman, E.; Avraham, R.; Benish, M.; Rosenne, E.; Ben-Eliyahu, S. Do stress responses promote leukemia progression? An animal study suggesting a role for epinephrine and prostaglandin-e2 through reduced nk activity. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Peng, Y.P.; Qiu, Y.H.; Jiang, J.L.; Wang, J.J. Effect of catecholamines on IL-2 production and NK cytotoxicity of rats in vitro. Acta Pharmacol. Sin. 2004, 25, 1354–1360. [Google Scholar]
- Yang, W.L.; Frucht, H. Cholinergic receptor up-regulates COX-2 expression and prostaglandin E2 production in colon cancer cells. Carcinogenesis 2000, 21, 1789–1793. [Google Scholar] [CrossRef]
- Wang, L.; Zhi, X.; Zhang, Q.; Wei, S.; Li, Z.; Zhou, J.; Jiang, J.; Zhu, Y.; Yang, L.; Xu, H.; et al. Muscarinic receptor M3 mediates cell proliferation induced by acetylcholine and contributes to apoptosis in gastric cancer. Tumor Biol. 2016, 37, 2105–2117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Shant, J.; Xie, G.; Cheng, K.; Gao, X.M.; Shiu, B.; Shah, N.; Drachenberg, C.B.; Heath, J.; Wess, J.; et al. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 2011, 32, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; Dai, X.; Gong, Q.; Bi, F. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pundavela, J.; Demont, Y.; Jobling, P.; Lincz, L.F.; Roselli, S.; Thorne, R.F.; Bond, D.; Bradshaw, R.A.; Walker, M.M.; Hondermarck, H. ProNGF correlates with Gleason score and is a potential driver of nerve infiltration in prostate cancer. Am. J. Pathol. 2014, 184, 3156–3162. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef]
- Pundavela, J.; Roselli, S.; Faulkner, S.; Attia, J.; Scott, R.J.; Thorne, R.F.; Forbes, J.F.; Bradshaw, R.A.; Walker, M.M.; Jobling, P.; et al. Nerve fibers infiltrate the tumor microenvironment and are associated with nerve growth factor production and lymph node invasion in breast cancer. Mol. Oncol. 2015, 9, 1626–1635. [Google Scholar] [CrossRef]
- Olar, A.; He, D.; Florentin, D.; Ding, Y.; Ayala, G. Biologic correlates and significance of axonogenesis in prostate cancer. Hum. Pathol. 2014, 45, 1358–1364. [Google Scholar] [CrossRef]
- Albo, D.; Akay, C.L.; Marshall, C.L.; Wilks, J.A.; Verstovsek, G.; Liu, H.; Agarwal, N.; Berger, D.H.; Ayala, G.E. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer 2011, 117, 4834–4845. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking nerve growth factor signaling reduces the neural invasion potential of pancreatic cancer cells. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.K.; Armaiz-Pena, G.N.; Nagaraja, A.S.; Sadaoui, N.C.; Ortiz, T.; Dood, R.; Ozcan, M.; Herder, D.M.; Haemerrle, M.; Gharpure, K.M.; et al. Sustained adrenergic signaling promotes intratumoral innervation through BDNF induction. Cancer Res. 2018, 78, 1701–2016. [Google Scholar] [CrossRef] [PubMed]
- Vanhecke, E.; Adriaenssens, E.; Verbeke, S.; Meignan, S.; Germain, E.; Berteaux, N.; Nurcombe, V.; Le Bourhis, X.; Hondermarck, H. Brain-derived neurotrophic factor and neurotrophin-4/5 are expressed in breast cancer and can be targeted to inhibit tumor cell survival. Clin. Cancer Res. 2011, 17, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yu, Y.; Song, Y.; Li, X.; Lan, D.; Zhang, P.; Xiao, Y.; Xing, Y. Activation of BDNF/TrkB pathway promotes prostate cancer progression via induction of epithelial-mesenchymal transition and anoikis resistance. FASEB J. 2020, 34, 9087–9101. [Google Scholar] [CrossRef]
- Choi, B.; Lee, E.J.; Shin, M.K.; Park, Y.S.; Ryu, M.H.; Kim, S.M.; Kim, E.Y.; Lee, H.K.; Chang, E.J. Upregulation of brain-derived neurotrophic factor in advanced gastric cancer contributes to bone metastatic osteolysis by inducing long pentraxin 3. Oncotarget 2016, 7, 55506–55517. [Google Scholar] [CrossRef]
- Chen, B.; Liang, Y.; He, Z.; An, Y.; Zhao, W.; Wu, J. Autocrine activity of BDNF induced by the STAT3 signaling pathway causes prolonged TrkB activation and promotes human non-small-cell lung cancer proliferation. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Bao, W.; Qiu, H.; Yang, T.; Luo, X.; Zhang, H.; Wan, X. Upregulation of TrkB Promotes Epithelial-Mesenchymal Transition and Anoikis Resistance in Endometrial Carcinoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ketterer, K.; Rao, S.; Friess, H.; Weiss, J.; Büchler, M.W.; Korc, M. Reverse Transcription-PCR Analysis of Laser-Captured Cells Points to Potential Paracrine and Autocrine Actions of Neurotrophins in Pancreatic Cancer. Clin. Cancer Res. 2003, 9, 5127–5136. [Google Scholar]
- Schlau, M.; Terheyden-Keighley, D.; Theis, V.; Mannherz, H.G.; Theiss, C. VEGF triggers the activation of cofilin and the Arp2/3 complex within the growth cone. Int. J. Mol. Sci. 2018, 19, 384. [Google Scholar] [CrossRef]
- Olbrich, L.; Foehring, D.; Happel, P.; Brand-Saberi, B.; Theiss, C. Fast rearrangement of the neuronal growth cone’s actin cytoskeleton following VEGF stimulation. Histochem. Cell Biol. 2013, 139, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Chédotal, A.; Kerjan, G.; Moreau-Fauvarque, C. The brain within the tumor: New roles for axon guidance molecules in cancers. Cell Death Differ. 2005, 12, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Rodrigues, G.; Hoshino, A.; Kenific, C.M.; Matei, I.R.; Steiner, L.; Freitas, D.; Kim, H.S.; Oxley, P.R.; Scandariato, I.; Casanova-Salas, I.; et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat. Cell Biol. 2019, 21, 1403–1412. [Google Scholar] [CrossRef]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.R.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef]
- Maeda, K.; Sasaki, H.; Ueda, S.; Miyamoto, S.; Terada, S.; Konishi, H.; Kogata, Y.; Ashihara, K.; Fujiwara, S.; Tanaka, Y.; et al. Serum exosomal microRNA-34a as a potential biomarker in epithelial ovarian cancer. J. Ovarian Res. 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Kurashige, J.; Kamohara, H.; Watanabe, M.; Tanaka, Y.; Kinoshita, K.; Saito, S.; Hiyoshi, Y.; Iwatsuki, M.; Baba, Y.; Baba, H. Serum microRNA-21 is a novel biomarker in patients with esophageal squamous cell carcinoma. J. Surg. Oncol. 2012, 106, 188–192. [Google Scholar] [CrossRef]
- Wei, J.; Gao, W.; Zhu, C.J.; Liu, Y.Q.; Mei, Z.; Cheng, T.; Shu, Y.Q. Identification of plasma microRNA-21 as a biomarker for early detection and chemosensitivity of non-small cell lung cancer. Chin. J. Cancer 2011, 30, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhuang, L.; Zhang, J.; Fan, J.; Luo, J.; Chen, H.; Wang, K.; Liu, L.; Chen, Z.; Meng, Z. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL. Mol. Oncol. 2013, 7, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Yang, L.F.; Zhu, Y.; Yao, X.D.; Zhang, S.L.; Dai, B.; Zhu, Y.P.; Shen, Y.J.; Shi, G.H.; Ye, D.W. Serum miRNA-21: Elevated levels in patients with metastatic hormone-refractory prostate cancer and potential predictive factor for the efficacy of docetaxel-based chemotherapy. Prostate 2011, 71, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, Z.; Zhao, Y.; Ding, Y.; Liu, H.; Xi, Y.; Xiong, W.; Li, G.; Lu, J.; Fodstad, O.; et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J. Biol. Chem. 2010, 285, 21496–21507. [Google Scholar] [CrossRef]
- Wang, H.; Tan, G.; Dong, L.; Cheng, L.; Li, K.; Wang, Z.; Luo, H. Circulating mir-125b as a marker predicting chemoresistance in breast cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS ONE 2012, 7, 1–5. [Google Scholar] [CrossRef]
- Ayuko Hoshino, A.; Sang Kim, H.; Bojmar, L.; Matei, I.R.; Jarnagin, W.R.; Lyden, D.; Hoshino, A.; Ennu Gyan, K.; Cioffi, M.; Hernandez, J.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers In Brief A comprehensive proteomic analysis of extracellular vesicles and particles (EVPs) from 426 human samples identifies pan-EVP markers, biomarkers for EVP isolation, for can. Cell 2020, 182, 1–18. [Google Scholar] [CrossRef]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas Analysis Reveals Distinct Extracellular RNA Cargo Types and Their Carriers Present across Human Biofluids. Cell 2019, 177, 463–477.e15. [Google Scholar] [CrossRef]
- Hong, C.S.; Funk, S.; Whiteside, T.L. Isolation of biologically active exosomes from plasma of patients with cancer. Methods Mol. Biol. 2017, 1633, 257–265. [Google Scholar] [CrossRef]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H. The clinical relevance of circulating, exosomal miRNAs as biomarkers for cancer. Expert Rev. Mol. Diagn. 2015, 15, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Nedaeinia, R.; Manian, M.; Jazayeri, M.H.; Ranjbar, M.; Salehi, R.; Sharifi, M.; Mohaghegh, F.; Goli, M.; Jahednia, S.H.; Avan, A.; et al. Circulating exosomes and exosomal microRNAs as biomarkers in gastrointestinal cancer. Cancer Gene Ther. 2017, 24, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Lu, T.-W.; Stolerman, L.M.; Tenner, B.; Yang, J.; Zhang, J.-F.; Rangamani, P.; Taylor, S.S.; Mehta, S.; Zhang, J. Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell 2020, 1–14. [Google Scholar] [CrossRef]
- Sapio, L.; Di Maiolo, F.; Illiano, M.; Esposito, A.; Chiosi, E.; Spina, A.; Naviglio, S. Targeting protein kinase a in cancer therapy: An update. EXCLI J. 2014, 13, 843–855. [Google Scholar] [CrossRef]
- Bucko, P.; Scott, J. Drugs that Regulate Local Cell Signaling: AKAP Targeting as a Therapeutic Option. Annu. Rev. Pharmacol. Toxicol. 2020, 61, 1–19. [Google Scholar] [CrossRef]
- Massimi, M.; Ragusa, F.; Cardarelli, S.; Giorgi, M. Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells 2019, 8, 1511. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Singh, P.; Alex, J.M.; Bast, F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: Novel treatment strategies for cancer. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [PubMed]
- Waldbillig, R.J.; LeRoith, D. Insulin receptors in the peripheral nervous system: A structural and functional analysis. Brain Res. 1987, 409, 215–220. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Nakamura, M.; Rikimaru, T.; Yano, T.; Moore, K.G.; Pula, P.J.; Schofield, B.H.; Dannenberg, A.M. Full-thickness human skin explants for testing the toxicity of topically applied chemicals. J. Invest. Dermatol. 1990, 95, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, R.L.; Falcinelli, M.; Flint, M.S. Stress and drug resistance in cancer. Cancer Drug Resist. 2019, 2, 773–786. [Google Scholar] [CrossRef]
- Lillemoe, K.D.; Cameron, J.L.; Kaufman, H.S.; Yeo, C.J.; Pitt, H.A.; Sauter, P.K. Chemical splanchnicectomy in patients with unresectable pancreatic cancer: A prospective randomized trial. Ann. Surg. 1993, 217, 447–457. [Google Scholar] [CrossRef]
- Demir, I.E.; Reyes, C.M.; Alrawashdeh, W.; Ceyhan, G.O.; Deborde, S.; Friess, H.; Görgülü, K.; Istvanffy, R.; Jungwirth, D.; Kuner, R.; et al. Clinically Actional Strategies for Studying Neural Influences in Cancer. Cancer Cell 2020, 1, 1–4. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Kamiya, A.; Hayama, Y.; Kato, S.; Shimomura, A.; Shimomura, T.; Irie, K.; Kaneko, R.; Yanagawa, Y.; Kobayashi, K.; Ochiya, T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 2019, 22, 1289–1305. [Google Scholar] [CrossRef]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylande, B.L. Adrenergic signaling: A targetable checkpoint limiting development of the antitumor immune response. Front. Immunol. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Melhem-Bertrandt, A.; Chavez-MacGregor, M.; Lei, X.; Brown, E.N.; Lee, R.T.; Meric-Bernstam, F.; Sood, A.K.; Conzen, S.D.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2011, 29, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.L.; Thaker, P.H.; Nick, A.M.; Ramondetta, L.M.; Kumar, S.; Urbauer, D.L.; Matsuo, K.; Squires, K.C.; Coleman, R.L.; Lutgendorf, S.K.; et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015, 121, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Grytli, H.H.; Fagerland, M.W.; Fosså, S.D.; Taskén, K.A. Association between use of β-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Halmos, B.; Wang, T.J.C.; Hei, T.K.; Cheng, S.K. Effects of β-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef]
- Rabben, H.-L.; Zhao, C.-M.; Hayakawa, Y.C.; Wang, T.; Chen, D.C. Vagotomy and Gastric Tumorigenesis. Curr. Neuropharmacol. 2016, 14, 967–972. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Molloy, N.H.; Read, D.E.; Gorman, A.M. Nerve growth factor in cancer cell death and survival. Cancers 2011, 3, 510–530. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Su, Y.; Pang, L.; Lu, Q.; Wang, Z.; Zhang, S.; Zheng, S.; Mao, J.; Zhu, Y. Inhibition of apoptosis by downregulation of hBex1, a novel mechanism, contributes to the chemoresistance of Bcr/Abl+ leukemic cells. Carcinogenesis 2009, 30, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Luan, Z.Y.; Han, F.; Chen, H.Q.; Liu, W.B.; Liu, J.Y.; Cao, J. Diagnostic and prognostic value of the BEX family in lung adenocarcinoma. Oncol. Lett. 2019, 18, 5523–5533. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xiao, Q.; Chen, H.; He, J.; Tan, Y.; Liu, Y.; Wang, Z.; Yang, Q.; Shen, X.; Huang, Y.; et al. BEX2 promotes tumor proliferation in colorectal cancer. Int. J. Biol. Sci. 2017, 13, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Miknyoczki, S.J.; Chang, H.; Klein-Szanto, A.; Dionne, C.A.; Ruggeri, B.A. The Trk tyrosine kinase inhibitor CEP-701 (KT-5555) exhibits significant antitumor efficacy in preclinical xenograft models of human pancreatic ductal adenocarcinoma. Clin. Cancer Res. 1999, 5, 2205–2212. [Google Scholar]
- Festuccia, C.; Muzi, P.; Gravina, G.L.; Millimaggi, D.; Speca, S.; Dolo, V.; Ricevuto, E.; Vicentini, C.; Bologna, M. Tyrosine kinase inhibitor CEP-701 blocks the NTRK1/NGF receptor and limits the invasive capability of prostate cancer cells in vitro. Int. J. Oncol. 2007, 30, 193–200. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Dalrymple, S.L.; Lamb, J.C.; Denmeade, S.R.; Isaacs, J.T.; Isaacs, J.T.; Miknyoczki, S.; Dionne, C.A. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin. Cancer Res. 2001, 7, 2237–2245. [Google Scholar]
- George, D.J.; Dionne, C.A.; Jani, J.; Angeles, T.; Murakata, C.; Lamb, J.; Isaacs, J.T. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555). Cancer Res. 1999, 59, 2395–2401. [Google Scholar]
- Jimenez-Andrade, J.M.; Ghilardi, J.R.; Castañeda-Corral, G.; Kuskowski, M.A.; Mantyh, P.W. Preventive or late administration of anti-NGF therapy attenuates tumor-induced nerve sprouting, neuroma formation, and cancer pain. Pain 2011, 152, 2564–2574. [Google Scholar] [CrossRef]
- Eibl, J.K.; Strasser, B.C.; Ross, G.M. Structural, biological, and pharmacological strategies for the inhibition of nerve growth factor. Neurochem. Int. 2012, 61, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Demir, I.E.; Tieftrunk, E.; Schorn, S.; Friess, H.; Ceyhan, G.O. Nerve growth factor & TrkA as novel therapeutic targets in cancer. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 37–50. [Google Scholar] [CrossRef]
- March, B.; Faulkner, S.; Jobling, P.; Steigler, A.; Blatt, A.; Denham, J.; Hondermarck, H. Tumour innervation and neurosignalling in prostate cancer. Nat. Rev. Urol. 2020, 17, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kappos, E.A.; Engels, P.E.; Tremp, M.; Sieber, P.K.; von Felten, S.; Madduri, S.; Meyer zu Schwabedissen, M.; Fischmann, A.; Schaefer, D.J.; Kalbermatten, D.F. Denervation leads to volume regression in breast cancer. J. Plast. Reconstr. Aesthetic Surg. 2018, 71, 833–839. [Google Scholar] [CrossRef]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412. [Google Scholar] [CrossRef]
- Coarfa, C.; Florentin, D.; Putluri, N.R.; Ding, Y.; Au, J.; He, D.; Ragheb, A.; Frolov, A.; Michailidis, G.; Lee, M.J.; et al. Influence of the neural microenvironment on prostate cancer. Prostate 2018, 78, 128–139. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Objective | Tumor of Interest | Trial ID |
|---|---|---|
| Document biomarker (protein kinase A) dynamics in cancer tissue | Colorectal cancer | NCT01012804 |
| Determine prognostic value of mu opioid receptor 1 expression/activation in cancer | Colorectal cancer | NCT04353882 |
| Determine prognostic value of SOX2 expression in colorectal cancer | Colorectal cancer | NCT01589900 |
| Assess the ability of common medications * to affect overall survival and disease-free survival | Pancreatic ductal adenocarcinoma | NCT04245644 |
| Objective | Anticancer Agent | Mechanism of Action | Phase | Tumor Targeted | Route of Administration | Trial ID |
|---|---|---|---|---|---|---|
| Assess the impact of bethanechol therapy on tumor activity | Bethanechol | Nonselective muscarinic activation | 1 | Pancreatic ductal adenocarcinoma | Oral | NCT03572283 |
| Identify a safe and pharmacologically active dose and regimen for VMD-928 monotherapy | VMD-928 | TrkA inhibition | 1 | Advanced solid tumors or lymphomas not responsive to available therapies * | Oral | NCT03556228 |
| Assess the safety and tolerability of entrectinib therapy | Entrectinib (RXDX-101) | TrkA, TrkB, TrkC, ROS1, ALK inhibition | 1 | Any locally advanced or metastatic cancer confirmed to be positive for NTRK1, NTRK2, NTRK3, ROS1, or ALK alterations | Oral | NCT02097810 |
| Measure therapeutic response in patients taking entrectinib | Entrectinib (RXDX-101) | TrkA, TrkB, TrkC, ROS1, ALK inhibition | 2 | Solid tumors that harbor a NTRK1/2/3, ROS1, or ALK gene fusion † | Oral | NCT02568267 |
| Determine the efficacy of carvedilol therapy | Carvedilol | Beta blockade | 2 | Prostate adenocarcinoma | Oral | NCT02944201 |
| Evaluate ADβR 2/PKA/BAD signal changes following treatment | Propranolol | Beta blockade | 2 | Prostate carcinoma | Oral | NCT03152786 |
| Evaluate the effects of propranolol and etodolac therapy on recurrence and biomarker expression | Propranolol and etodolac | Beta blockade and COX2 inhibition | 2 | Pancreatic cancers | Oral | NCT03838029 |
| Obtain the data needed to calculate sample size for a larger controlled trial | Botulinum toxin | Acetylcholine release inhibition | 2 | Stomach cancer | Injection by gastroscopy | NCT01822210 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunt, P.J.; Kabotyanski, K.E.; Calin, G.A.; Xie, T.; Myers, J.N.; Amit, M. Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance. Cancers 2020, 12, 3741. https://doi.org/10.3390/cancers12123741
Hunt PJ, Kabotyanski KE, Calin GA, Xie T, Myers JN, Amit M. Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance. Cancers. 2020; 12(12):3741. https://doi.org/10.3390/cancers12123741
Chicago/Turabian StyleHunt, Patrick J., Katherine E. Kabotyanski, George A. Calin, Tongxin Xie, Jeffrey N. Myers, and Moran Amit. 2020. "Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance" Cancers 12, no. 12: 3741. https://doi.org/10.3390/cancers12123741
APA StyleHunt, P. J., Kabotyanski, K. E., Calin, G. A., Xie, T., Myers, J. N., & Amit, M. (2020). Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance. Cancers, 12(12), 3741. https://doi.org/10.3390/cancers12123741