Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity

 , and
, and
Abstract
:Simple Summary
Abstract
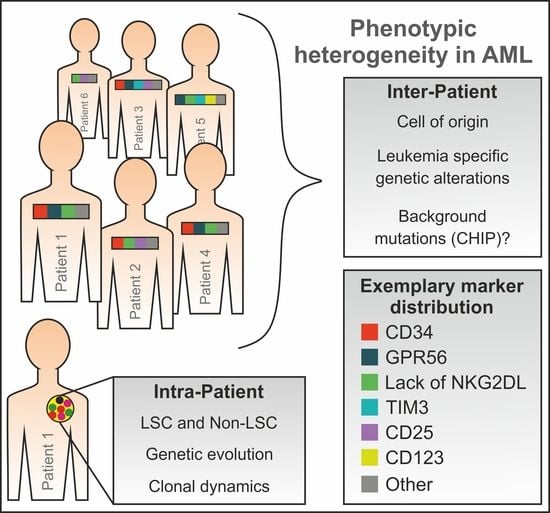
1. Introduction
2. Leukemic Stem Cells and Healthy Stem/Progenitor Cells
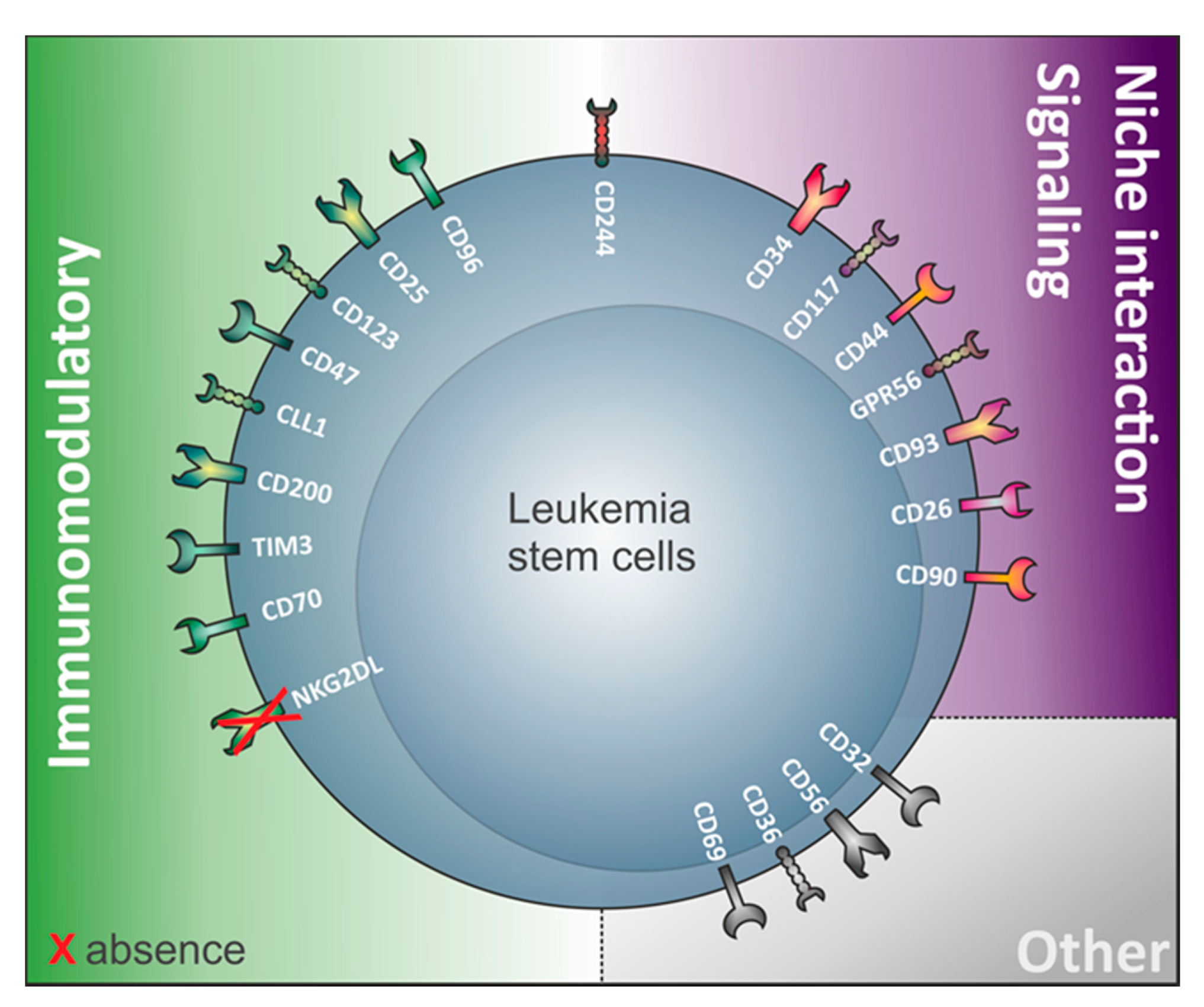
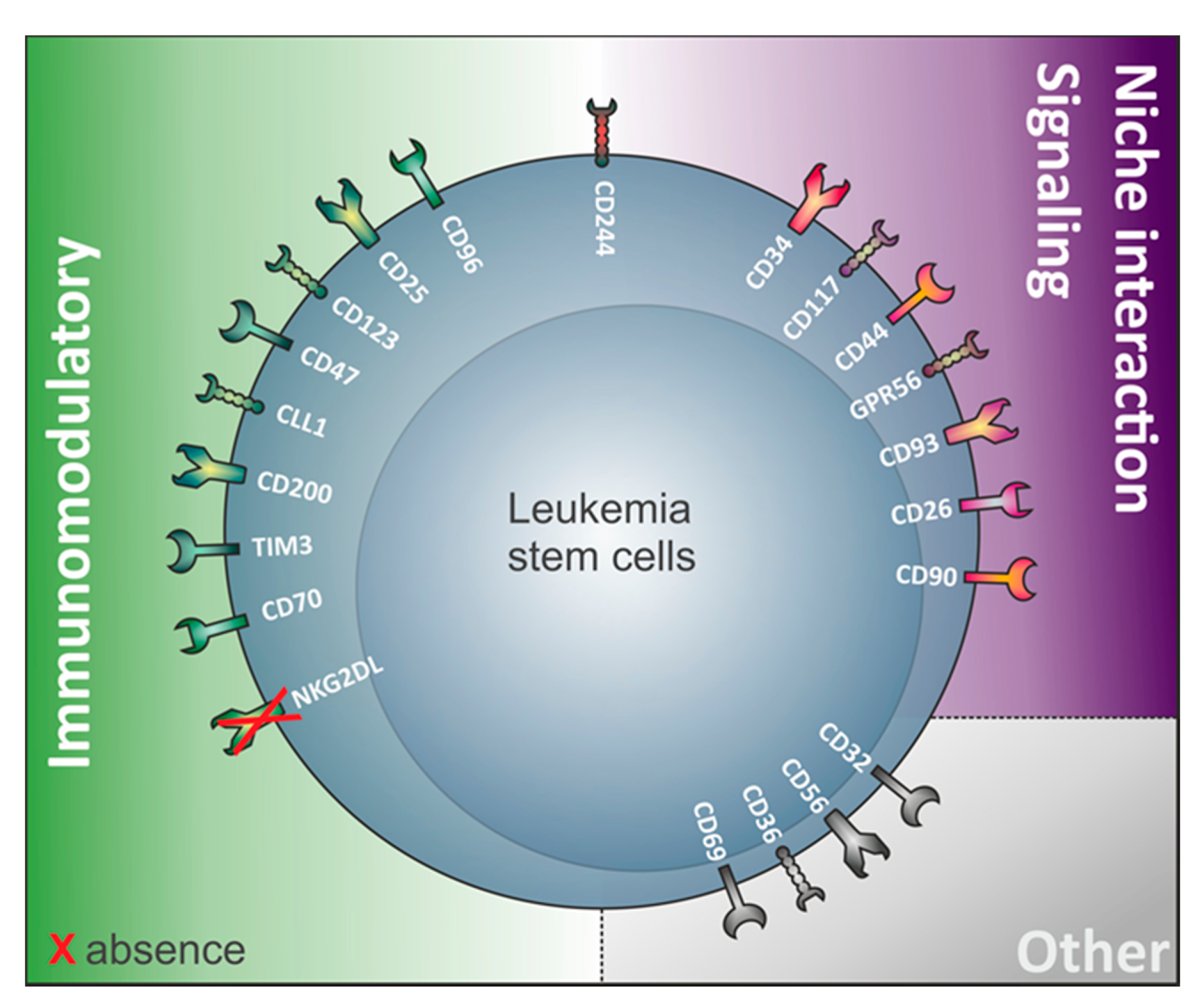
3. The Relevance of Immunomodulatory Proteins for LSC Detection
4. LSC Surface Markers in CD34 Expressing Compared to CD34 Non-Expressing AML
4.1. CD34 Expressing AML Contain CD34+ LSC
4.2. CD34 Non-Expressing AML and Their LSC
4.3. Review of Markers Capturing LSC in AML Samples Regardless of Their CD34 Expression
4.3.1. Absence of NKG2D Ligands
4.3.2. GPR56
4.3.3. CD200
5. Phenotypic LSC Evolution and Intra-Patient Heterogeneity
6. Association between the Genetic Background and the LSC Phenotype in AML
6.1. GPR56
6.2. CD93
6.3. CD26
7. Therapeutic Targeting of LSC
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Bahr, C.; Correia, N.C.; Trumpp, A. Stem cells make leukemia grow again. EMBO J. 2017, 36, 2667–2669. [Google Scholar] [CrossRef]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Wang, X.; Huang, S.; Chen, J.L. Understanding of leukemic stem cells and their clinical implications. Mol. Cancer 2017, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Guilak, F.; Cohen, D.M.; Estes, B.T.; Gimble, J.M.; Liedtke, W.; Chen, C.S. Control of Stem Cell Fate by Physical Interactions with the Extracellular Matrix. Cell Stem Cell 2009, 5, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Hoggatt, J.; Kfoury, Y.; Scadden, D.T. Hematopoietic Stem Cell Niche in Health and Disease. Annu. Rev. Pathol. 2016, 11, 555–581. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aguilera, A.; Méndez-Ferrer, S. The hematopoietic stem-cell niche in health and leukemia. Cell. Mol. Life Sci. 2017, 74, 579–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passegué, E.; Jamieson, C.H.M.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Engelhardt, M.; Lübbert, M.; Guo, Y. CD34+ or CD34−: Which is the more primitive? Leukemia 2002, 16, 1603–1608. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.S.; McNagny, K.M. Erratum: Novel functions of the CD34 family. J. Cell Sci. 2008, 121, 3683–3692. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Saito, Y.; Morishita, K. Maintenance of leukemic and normal hematopoietic stem cells in bone marrow niches by EVI1-regulated GPR56. Rinsho Ketsueki 2015, 56, 375–383. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef]
- Zagozdzon, R.; Golab, J. Cancer stem cells in haematological malignancies. Wspolczesna Onkol. 2015, 19, A1–A6. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic Detection of Measurable Residual (Stem Cell) Disease Using LAIP Approach in Acute Myeloid Leukemia. Curr. Protoc. Cytom. 2019, 91, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J. Stem Cells 2014, 6, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Thomas, R.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of target expression profiles in CD34+/CD38− and CD34+/CD38− stem and progenitor cells in AML and CML. Blood Adv. 2020, 4, 5118–5132. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.M.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia 2016, 30, 439–446. [Google Scholar] [CrossRef]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of age: CD96 emerges as modulator of immune responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An activation marker and activation limiter of innate immune cells. Front. Immunol. 2013, 4, 449. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.S.J.; Willment, J.A.; Pyz, E.; Dennehy, K.M.; Reid, D.M.; Dri, P.; Gordon, S.; Wong, S.Y.C.; Brown, G.D. Human MICL (CLEC12A) is differentially glycosylated and is down-regulated following cellular activation. Eur. J. Immunol. 2006, 36, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Anania, J.C.; Chenoweth, A.M.; Wines, B.D.; MarkHogarth, P. The human FcγRII (CD32) family of leukocyte FCR in health and disease. Front. Immunol. 2019, 10, 464. [Google Scholar] [CrossRef]
- Triplett, T.A.; Curti, B.D.; Bonafede, P.R.; Miller, W.L.; Walker, E.B.; Weinberg, A.D. Defining a functionally distinct subset of human memory CD4+ T cells that are CD25POS and FOXP3NEG. Eur. J. Immunol. 2012, 42, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Guthridge, M.A.; Stomski, F.C.; Thomas, D.; Woodcock, J.M.; Bagley, C.J.; Berndt, M.C.; Lopez, A.F. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells 1998, 16, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J. Clin. Investig. 2011, 121, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.J.; Doondeea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically distinct leukemic stem cells in human CD34− acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J. Exp. Med. 2016, 213, 1513–1535. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Vargaftig, J.; Miraki-Moud, F.; Griessinger, E.; Sharrock, K.; Luke, T.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34− fraction. Blood 2010, 115, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Kersten, B.; Valkering, M.; Wouters, R.; van Amerongen, R.; Hanekamp, D.; Kwidama, Z.; Valk, P.; Ossenkoppele, G.; Zeijlemaker, W.; Kaspers, G.; et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br. J. Haematol. 2016, 173, 219–235. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Holmes, N. CD45: All is not yet crystal clear. Immunology 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Vergez, F.; Green, A.S.; Tamburini, J.; Sarry, J.E.; Gaillard, B.; Cornillet-Lefebvre, P.; Pannetier, M.; Neyret, A.; Chapuis, N.; Ifrah, N.; et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: A Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica 2011, 96, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Vergez, F.; Nicolau-Travers, M.L.; Bertoli, S.; Rieu, J.B.; Tavitian, S.; Bories, P.; Luquet, I.; Mas, V.; Largeaud, L.; Sarry, A.; et al. CD34+CD38−CD123+ Leukemic Stem Cell Frequency Predicts Outcome in Older Acute Myeloid Leukemia Patients Treated by Intensive Chemotherapy but Not Hypomethylating Agents. Cancers 2020, 12, 1174. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Pinto, A.D.; Al-Zadjali, S. CD34+CD38-CD123+ Cells Are Present in Virtually All Acute Myeloid Leukaemia Blasts: A Promising Single Unique Phenotype for Minimal Residual Disease Detection. Acta Haematol. 2017, 138, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bras, A.E.; de Haas, V.; van Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; te Marvelde, J.G.; Zwaan, C.M.; van Dongen, J.J.M.; Leusen, J.H.W.; van der Velden, V.H.J. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytom. Part B Clin. Cytom. 2019, 96, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Paprocka, M.; Bielawska-Pohl, A.; Rossowska, J.; Krawczenko, A.; Duś, D.; Kiełbiński, M.; Haus, O.; Podolak-Dawidziak, M.; Kuliczkowski, K. MRP1 protein expression in leukemic stem cells as a negative prognostic marker in acute myeloid leukemia patients. Eur. J. Haematol. 2017, 99, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Touzet, L.; Dumezy, F.; Roumier, C.; Berthon, C.; Bories, C.; Quesnel, B.; Preudhomme, C.; Boyer, T. CD9 in acute myeloid leukemia: Prognostic role and usefulness to target leukemic stem cells. Cancer Med. 2019, 8, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.L.; Taki, T.; Adachi, M.; Yagita, M.; Sawada, S.; Takabayashi, A.; Inufusa, H.; Yoshie, O.; Miyake, M. MRP-1/CD9 and KAI1/CD82 expression in normal and various cancer tissues. Int. J. Oncol. 1997, 11, 1045–1051. [Google Scholar] [CrossRef]
- Wang, V.M.Y.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Thin, M.Z.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef]
- Brendel, C.; Mohr, B.; Schimmelpfennig, C.; Muller, J.; Bornhauser, M.; Schmidt, M.; Ritter, M.; Ehninger, G.; Neubauer, A. Detection of cytogenetic aberrations both in CD90 (Thy-1)-positive and (Thy-1)-negative stem cell (CD34) subfractions of patients with acute and chronic myeloid leukemias. Leukemia 1999, 13, 1770–1775. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Weissman, I.L.; Akashi, K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 7521–7526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, A.; Hogge, D.E.; Ailles, L.E.; Lansdorp, P.M.; Sutherland, H.J. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 1997, 89, 3104–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, W.; Kay, R.; Cutler, R.L.; Lansdorp, P.M. Expression of Thy-1 on human hematopoietic progenitor cells. J. Exp. Med. 1993, 177, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Daga, S.; Rosenberger, A.; Quehenberger, F.; Krisper, N.; Prietl, B.; Reinisch, A.; Zebisch, A.; Sill, H.; Wolfler, A. High GPR56 surface expression correlates with a leukemic stem cell gene signature in CD34-positive AML. Cancer Med. 2019, 8, 1771–1778. [Google Scholar] [CrossRef]
- Larsen, H.O.; Roug, A.S.; Just, T.; Brown, G.D.; Hokland, P. Expression of the hMICL in acute myeloid leukemia-a highly reliable disease marker at diagnosis and during follow-up. Cytom. B Clin. Cytom. 2012, 82, 3–8. [Google Scholar] [CrossRef]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- Brosseau, C.; Colas, L.; Magnan, A.; Brouard, S. CD9 tetraspanin: A new pathway for the regulation of inflammation? Front. Immunol. 2018, 9, 2316. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra19. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, Y.; Miwa, H.; Arakawa, R.; Tawara, I.; Ohishi, K.; Masuya, M.; Nakase, K.; Katayama, N. Expression of CD25 fluctuates in the leukemia-initiating cell population of CD25-positive AML. PLoS ONE 2018, 13, e0209295. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Wagner, L.; Stephan, M.; von Hörsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-L.; Yuan, J.-Y.; Zhang, J.-W.; Zhang, X.-H.; Wang, R.-X. Differential gene expression in human hematopoietic stem cells specified toward erythroid, megakaryocytic, and granulocytic lineage. J. Leukoc. Biol. 2007, 82, 986–1002. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Sachs, K.; Sarver, A.L.; Noble-Orcutt, K.E.; LaRue, R.S.; Antony, M.L.; Chang, D.; Lee, Y.; Navis, C.M.; Hillesheim, A.L.; Nykaza, I.R.; et al. Single-cell gene expression analyses reveal distinct self-renewing and proliferating subsets in the leukemia stem cell compartment in acute myeloid leukemia. Cancer Res. 2020, 80, 458–470. [Google Scholar] [CrossRef]
- Keyhani, A.; Huh, Y.O.; Jendiroba, D.; Pagliaro, L.; Cortez, J.; Pierce, S.; Pearlman, M.; Estey, E.; Kantarjian, H.; Freireich, E.J. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk. Res. 2000, 24, 153–159. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Bendall, L.J.; Bradstock, K.F.; Gottlieb, D.J. Expression of CD44 variant exons in acute myeloid leukemia is more common and more complex than that observed in normal blood, bone marrow or CD34+ cells. Leukemia 2000, 14, 1239–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sick, E.; Jeanne, A.; Schneider, C.; Dedieu, S.; Takeda, K.; Martiny, L. CD47 update: A multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 2012, 167, 1415–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the immune system: More than a marker for cytotoxicity? Front. Immunol. 2017, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- Sasca, D.; Szybinski, J.; Schüler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.M.; Gebhardt, W.H.; et al. NCAM1 (CD56) promotes leukemogenesis and confers drug resistance in AML. Blood 2019, 133, 2305–2319. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Riether, C.; Schürch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Riether, C.; Schürch, C.M.; Bührer, E.D.; Hinterbrandner, M.; Huguenin, A.L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J. Exp. Med. 2017, 214, 359–380. [Google Scholar] [CrossRef]
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005, 17, 275–281. [Google Scholar] [CrossRef]
- Buccisano, F.; Rossi, F.M.; Venditti, A.; Del Poeta, G.; Cox, M.C.; Abbruzzese, E.; Rupolo, M.; Berretta, M.; Degan, M.; Russo, S.; et al. CD90/Thy-1 is preferentially expressed on blast cells of high risk acute myeloid leukaemias. Br. J. Haematol. 2004, 125, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Kisselbach, L.; Merges, M.; Bossie, A.; Boyd, A. CD90 expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology 2009, 59, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Bohlson, S.; Greenlee, M.; Sullivan, S. CD93 and Related Family Members: Their Role in Innate Immunity. Curr. Drug Targets 2008, 9, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Liedtke, M.; Gentles, A.J.; Cleary, M.L. CD93 marks a non-quiescent human leukemia stem cell population and is required for development of MLL-rearranged acute myeloid leukemia. Cell Stem Cell 2015, 17, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumide, K.; Matsuoka, Y.; Kawamura, H.; Nakatsuka, R.; Fujioka, T.; Asano, H.; Takihara, Y.; Sonoda, Y. A revised road map for the commitment of human cord blood CD34-negative hematopoietic stem cells. Nat. Commun. 2018, 9, 2202. [Google Scholar] [CrossRef] [PubMed]
- Al-Fatlawi, H.; Musa, R. Evaluation of CD96 and CD123 in CD34+ leukemic stem cells in acute myeloid leukemia patients and their relation to response to induction therapy. Iraqi J. Hematol. 2016, 5, 161–166. [Google Scholar] [CrossRef]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013. [Google Scholar] [CrossRef] [Green Version]
- Sperling, C.; Schwartz, S.; Büchner, T.; Thiel, E.; Ludwig, W.D. Expression of the stem cell factor receptor C-KIT (CD117) in acute leukemias. Haematologica 1997, 82, 617–621. [Google Scholar]
- Geissler, E.N.; Liao, M.; Brook, J.D.; Martin, F.H.; Zsebo, K.M.; Housman, D.E.; Galli, S.J. Stem cell factor (SCF), a novel hematopoietic growth factor and ligand for c-kit tyrosine kinase receptor, maps on human chromosome 12 between 12q14.3 and 12qter. Somat Cell Mol. Genet. 1991, 17, 207–214. [Google Scholar] [CrossRef]
- Wells, S.J.; Bray, R.A.; Stempora, L.L.; Farhi, D.C. CD117/CD34 expression in leukemic blasts. Am. J. Clin. Pathol. 1996, 106, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, C.; Liu, F. CD200-CD200R signaling and diseases: A potential therapeutic target? Int. J. Physiol. Pathophysiol. Pharmcol. 2019, 11, 297–309. [Google Scholar]
- Ho, J.M.; Dobson, S.M.; Voisin, V.; McLeod, J.; Kennedy, J.A.; Mitchell, A.; Jin, L.; Eppert, K.; Bader, G.; Minden, M.D.; et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020, 4, 5402–5413. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, X.; Chen, C.; Zhu, J.; Yu, Z.; Xie, J.; Xie, L.; Bai, H.; Zhang, Y.; Fang, X.; et al. CD244 maintains the proliferation ability of leukemia initiating cells through SHP-2/p27kip1 signaling. Haematologica 2017, 102, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front. Immunol. 2018, 9, 2809. [Google Scholar] [CrossRef] [PubMed]
- Pabst, C.; Bergeron, A.; Lavallée, V.P.; Yeh, J.; Gendron, P.; Norddahl, G.L.; Krosl, J.; Boivin, I.; Deneault, E.; Simard, J.; et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016, 127, 2018–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaimani Kartalaei, P.; Yamada-Inagawa, T.; Vink, C.S.; de Pater, E.; van der Linden, R.; Marks-Bluth, J.; van der Sloot, A.; van den Hout, M.; Yokomizo, T.; van Schaick-Solerno, M.L.; et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J. Exp. Med. 2015, 212, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.Y.; Lin, H.H. The activation and signaling mechanisms of GPR56/ADGRG1 in melanoma cell. Front. Oncol. 2018, 8, 304. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and its ligands: “One for all, all for one”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Piao, X.; Hill, R.S.; Bodell, A.; Chang, B.S.; Basel-Vanagaite, L.; Straussberg, R.; Dobyns, W.B.; Qasrawi, B.; Winter, R.M.; Innes, A.M.; et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004, 303, 2033–2036. [Google Scholar] [CrossRef] [Green Version]
- Saha, H.R.; Kaneda-Nakashima, K.; Shimosaki, S.; Suekane, A.; Sarkar, B.; Saito, Y.; Ogoh, H.; Nakahata, S.; Inoue, K.; Watanabe, T.; et al. Suppression of GPR56 expression by pyrrole-imidazole polyamide represents a novel therapeutic drug for AML with high EVI1 expression. Sci. Rep. 2018, 8, 13741. [Google Scholar] [CrossRef] [Green Version]
- Daria, D.; Kirsten, N.; Muranyi, A.; Mulaw, M.; Ihme, S.; Kechter, A.; Hollnagel, M.; Bullinger, L.; Döhner, K.; Döhner, H.; et al. GPR56 contributes to the development of acute myeloid leukemia in mice. Leukemia 2016, 30, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Hills, R.K.; Wang, E.C.; Burnett, A.K.; Man, S.; Darley, R.L.; Tonks, A. Expression of CD200 on AML blasts directly suppresses memory T-cell function. Leukemia 2012, 26, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Wang, E.C.; Man, S.; Hills, R.K.; Burnett, A.K.; Tonks, A.; Darley, R.L. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011, 25, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rhenen, A.; van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Stigter-van Walsum, M.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Byun, J.M.; Shin, D.Y.; Yoon, S.S.; Koh, Y.; Hong, J.; Kim, I.; Lee, C.; Yoo, H.; Yun, H.; et al. Leukemic stem cell phenotype is associated with mutational profile in acute myeloid leukemia. Korean J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Khan, N.; Hills, R.K.; Virgo, P.; Couzens, S.; Clark, N.; Gilkes, A.; Richardson, P.; Knapper, S.; Grimwade, D.; Russell, N.H.; et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017, 31, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Béné, M.-C.; Porwit, A. Examples of Immunophenotypic Features in Various Categories of Acute Leukaemia. In Multiparameter Flow Cytometry in the Diagnosis of Hematologic Malignancies; Porwit, A., Béné, M.C., Eds.; Cambridge University Press: Cambridge, UK, 2018; pp. 75–88. [Google Scholar]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ Immunophenotype Identifies FLT3-Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Reyes-Palomares, A.; He, L.; Bergeron, A.; Lavallée, V.P.; Lemieux, S.; Gendron, P.; Rohde, C.; Xia, J.; Jagdhane, P.; et al. Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3-ITD triple-mutated AML. Blood 2019, 134, 263–276. [Google Scholar] [CrossRef]
- Kinstrie, R.; Horne, G.A.; Morrison, H.; Irvine, D.; Munje, C.; Castañeda, E.G.; Moka, H.A.; Dunn, K.; Cassels, J.E.; Parry, N.; et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia 2020, 34, 1613–1625. [Google Scholar] [CrossRef] [Green Version]
- Taussig, D.C.; Pearce, D.J.; Simpson, C.; Rohatiner, A.Z.; Lister, T.A.; Kelly, G.; Luongo, J.L.; Danet-Desnoyers, G.A.; Bonnet, D. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef] [Green Version]
- Hauswirth, A.W.; Florian, S.; Printz, D.; Sotlar, K.; Krauth, M.T.; Fritsch, G.; Schernthaner, G.H.; Wacheck, V.; Selzer, E.; Sperr, W.R.; et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur. J. Clin. Investig. 2007, 37, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kügler, M.; Stein, C.; Kellner, C.; Mentz, K.; Saul, D.; Schwenkert, M.; Schubert, I.; Singer, H.; Oduncu, F.; Stockmeyer, B.; et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br. J. Haematol. 2010, 150, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Pizzitola, I.; Anjos-Afonso, F.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richte, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.; Seedhouse, C.; Mony, U.; Grundy, M.; Russell, N.H.; Pallis, M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia 2010, 24, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A.; et al. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Selby, C.; Yacko, L.R.; Glode, A.E. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pract. Oncol. 2019, 10, 68–82. [Google Scholar]
- Egan, P.C.; Reagan, J.L. The return of gemtuzumab ozogamicin: A humanized anti-CD33 monoclonal antibody-drug conjugate for the treatment of newly diagnosed acute myeloid leukemia. OncoTargets Ther. 2018, 11, 8265–8272. [Google Scholar] [CrossRef] [Green Version]
- Theocharides, A.P.; Jin, L.; Cheng, P.Y.; Prasolava, T.K.; Malko, A.V.; Ho, J.M.; Poeppl, A.G.; van Rooijen, N.; Minden, M.D.; Danska, J.S.; et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 2012, 209, 1883–1899. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce, L.P.; Fenn, N.C.; Moritz, N.; Krupka, C.; Kozik, J.H.; Lauber, K.; Subklewe, M.; Hopfner, K.P. SIRPalpha-antibody fusion proteins stimulate phagocytosis and promote elimination of acute myeloid leukemia cells. Oncotarget 2017, 8, 11284–11301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134 (Suppl. 1), 569. [Google Scholar] [CrossRef]
- Yan, B.; Chen, Q.; Shimada, K.; Tang, M.; Li, H.; Gurumurthy, A.; Khoury, J.D.; Xu, B.; Huang, S.; Qiu, Y. Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia. Leukemia 2019, 33, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Eladl, E.; Tremblay-LeMay, R.; Rastgoo, N.; Musani, R.; Chen, W.; Liu, A.; Chang, H. Role of CD47 in Hematological Malignancies. J. Hematol. Oncol. 2020, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.T.; Murphy, M.F. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Stauder, R.; Curt, F.; Oliferenko, S.; Marie, J.P.; Proctor, S.; Jasmin, C. A strong expression of CD44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood 1998, 91, 3401–3413. [Google Scholar]
- Charrad, R.S.; Li, Y.; Delpech, B.; Balitrand, N.; Clay, D.; Jasmin, C.; Chomienne, C.; Smadja-Joffe, F. Ligation of the CD44 adhesion molecule reverses blockage of differentiation in human acute myeloid leukemia. Nat. Med. 1999, 5, 669–676. [Google Scholar] [CrossRef]
- Charrad, R.S.; Gadhoum, Z.; Qi, J.; Glachant, A.; Allouche, M.; Jasmin, C.; Chomienne, C.; Smadja-Joffe, F. Effects of anti-CD44 monoclonal antibodies on differentiation and apoptosis of human myeloid leukemia cell lines. Blood 2002, 99, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Gadhoum, Z.; Leibovitch, M.P.; Oi, J.; Dumenil, D.; Durand, L.; Leibovitch, S.; Smadja-Joffe, F. CD44: A new means to inhibit acute myeloid leukemia cell proliferation via p27Kip1. Blood 2004, 103, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Liao, X.; Zhou, L.; Wu, L.; Feng, Y.; Han, Z.C. HI44a, an anti-CD44 monoclonal antibody, induces differentiation and apoptosis of human acute myeloid leukemia cells. Leuk. Res. 2004, 28, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Delaunay, J.; Martinelli, G.; Fiedler, W.; Raffoux, E.; Prebet, T.; Gomez-Roca, C.; Papayannidis, C.; Kebenko, M.; Paschka, P.; et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 2016, 7, 32532–32542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; Aly, S.S.; Rayan, A.; El-Badawy, O.; Fattah, M.A.; Ali, A.M.; ElBadre, H.M.; Hetta, H.F. Survival outcomes of CD34+CD38−LSCs and their expression of CD123 in adult AML patients. Oncotarget 2018, 9, 34056–34065. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a therapeutic target in the treatment of hematological malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sallman, D.A.; Sweet, K.L.; Rizzieri, D.A.; Sayre, P.H.; Advani, A.S.; Emadi, A.; Wieduwilt, M.J.; et al. Flotetuzumab, an Investigational CD123 x CD3 Bispecific Dart® Protein, in Salvage Therapy for Primary Refractory and Early Relapsed Acute Myeloid Leukemia (AML) Patients. Blood 2019, 134 (Suppl. 1), 733. [Google Scholar] [CrossRef]
- Wei, A.H.; Fong, C.Y.; Montesinos, P.; Calbacho, M.; Gil, J.S.; Perez De Oteyza, J.; Rowe, J.M.; Wolach, O.; Ofran, Y.; Moshe, Y.; et al. A Phase 1 Study of Flotetuzumab, a CD123 x CD3 DART® Protein, Combined with MGA012, an Anti-PD-1 Antibody, in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2019, 134 (Suppl. 1), 2662. [Google Scholar] [CrossRef]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130 (Suppl. 1), 811. [Google Scholar] [CrossRef]
- Loff, S.; Dietrich, J.; Meyer, J.E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Oncolytics 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.R.; Crimmins, M.A.; Yetz-Aldape, J.; Kriz, R.; Kelleher, K.; Herrmann, S. The cloning of CD70 and its identification as the ligand for CD27. J. Immunol. 1994, 152, 1756–1761. [Google Scholar] [PubMed]
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H. David Francisco, Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat. Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Yabushita, T.; Satake, H.; Maruoka, H.; Morita, M.; Katoh, D.; Shimomura, Y.; Yoshioka, S.; Morimoto, T.; Ishikawa, T. Expression of multiple leukemic stem cell markers is associated with poor prognosis in de novo acute myeloid leukemia. Leuk. Lymphoma 2018, 59, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.M.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34+CD38− stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonardi, F.; Fusetti, F.; Deelen, P.; van Gosliga, D.; Vellenga, E.; Schuringa, J.J. A proteomics and transcriptomics approach to identify leukemic stem cell (LSC) markers. Mol. Cell. Proteom. 2013, 12, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Raffel, A.S.; Klimmeck, D.; Falcone, M.; Demir, A.; Straße, W. Quantitative Proteomics Reveals Specific Metabolic Features of Acute Myeloid Leukemia Stem Cells; Heidelberg Institute for Stem Cell Technology and Experimenta: Heidelberg, Germany, 2020. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Antigen | Percentage of AML Patients Expressing the Marker | Expression on Non-LSC | Expression on HSC | Expression on Other Healthy Blood Cells | Function in Healthy Conditions | References |
|---|---|---|---|---|---|---|
| CLL-1 | 92 | Yes | No | Monocytes, granulocytes, CMP, GMP | Modulates the activation state of cells during inflammation processes | Bakker et al. 2004 [57] Jiang et al. 2018 [58] Daga et al. 2019 [55] Marshall et al. 2006 [29] |
| CD9 | 40 | Yes | No | Monocytes, macrophages, granulocytes, DC, endothelial cells, B, T, and NK cells | Cell migration, adhesion, activation, | Brosseau et al. 2018 [59] Touzet et al. 2019 [47] Paprocka et al. 2017 [46] |
| CD25 | 10–25 | Yes | No | T cells and regulatory T cells | Important role for T cells survival | Saito et al. 2010 [60] Kageyama et al. 2018 [61] Triplett et al. 2012 [31] |
| CD26 | N.D | Yes | No | T, B, NK, and myeloid cells | T cell activation and proliferation, cell adhesion, metabolism | Herrmann et al. 2020 [25] Klemann et al. 2016 [62] |
| CD32 | 35 | Yes | No | Monocytes, B and T cells | Immune cell activation | Saito et al. 2010 [60] Anania et al. 2019 [30] |
| CD33 | 88 | Yes | Yes | Myeloid cells, lymphocytes, NK cells, MPP, GMP, MEP | Modulates inflammatory and immune responses by reducing tyrosine kinase dependent pathways | Ehninger et al. 2014 [63] Liu et al. 2007 [64] Laszlo et al. 2014 [65] Haubner et al. 2017 [24] |
| CD34 | 70 | Yes | Yes | Mast cells, eosinophils, neurons, fibrocytes | Regulates cell differentiation, adhesion, trafficking and proliferation | Quek et al. 2016 [36] Engelhardt et al. 2002 [16] Nielsen et al. 2008 [17] |
| CD36 | N.D | Yes | No | Platelets, monocytes, adipocytes | Fatty acid uptake, angiogenesis, PRR recognition | Silverstein et al. 2009 [66] Sachs et al. 2020 [67] Herrmann et al. 2020 [25] |
| CD38 | 5–55 (FAB subtypes) | Yes | No | T and B cells, monocytes, NK, granulocytes, platelets, red blood cells | Regulates calcium levels and NAD+ homeostasis | Hogan et al. 2019 [38] Sarry et al. 2011 [35] Goardon et al. 2011 [40] Keyhani et al. 2000 [68] |
| CD44 | N.D | Yes | Yes | T cells, mesenchymal cells, ectodermal cells, neuron-like cells | Cell adhesion molecule, cellular signaling | Ponta et al. 2003 [69] Jin et al. 2006 [70] Bendall et al. 2000 [71] Herrmann et al. 2020 [25] |
| CD45RA | N.D | Yes | Yes | T and B cells | CD45 isoform, cell signaling | Kersten et al. 2016 [39] Goardon et al. 2011 [40] Sarry et al. 2011 [35] Holmes 2006 [41] |
| CD47 | N.D | Yes | Yes | Various healthy cells | “don’t eat me” signal on cells in order to prevent inappropriate phagocytosis | Majeti et al. 2009 [34] Jaiswal et al. 2009 [33] Sick et al. 2012 [72] |
| CD56 | Up to 20 | Yes | No | DC, T and NK cells | Linked to NK cytotoxicity | Van Acker et al. 2017 [73] Sasca et al. 2019 [74] Herrmann et al. 2020 [25] |
| CD69 | N.D | N.D | No | T cells | T cell differentiation, tissue retention, and metabolic reprogramming | Cibrián et al. 2017 [75] Sachs et al. 2020 [67] Herrmann et al. 2020 [25] |
| CD70 | N.D | Yes | No | DC | T and B cell activation | Riether et al. 2015 [76] Riether et al. 2017 [77] Borst et al. 2005 [78] |
| CD90 | 40 (in elderly patients) | Yes | Yes | Fibroblasts, neurons, endothelial cells | Maintenance of HSC, cell adhesion, matrix adhesion | Buccisano et al. 2004 [79] Blair et al. 1997 [52] Brendel et al. 1999 [50] Kisselbach et al. 2009 [80] Craig et al. 1993 [53] |
| CD93 | N.D | N.D | No (only on CD34-HSC) | Myeloid and endothelial cells | Mechanism in innate host defense | Bohlson et al. 2008 [81] Iwasaki et al. 2015 [82] Sumide et al. 2018 [83] |
| CD96 | 27 | Yes | Only 5% | T and NK cells | Inhibits NK and T cells | Fatlawi et al. 2016 [84] Georgiev et al. 2018 [27] Hosen et al. 2007 [85] |
| CD117 | 87 | Yes | Yes | GMP | Promotes HSC growth by binding the stem cell factor | Sperling et al. 1997 [86] Geissler et al. 1991 [87] Quek et al. 2016 [36] Wells et al. 1996 [88] |
| CD123 | 97 | Yes | No | Basophils, plasmacytoid DC | Proliferation, survival, activation, and differentiation by binding respective ligand | Yu et al. 2016 [88] Guthridge et al. 1998 [32] Bras et al. 2019 [45] Haubner et al. 2019 [24] Al-Mawali et al. 2017 [44] |
| CD200 | N.D | Yes | Yes | Myeloid, T and B cells | Immunoregulatory molecule | Ngwa et al. 2019 [89] Ho et al. 2020 [90] |
| CD244 | N.D | Yes | Yes | GMP, HSPC, granulocytes, monocytes, DC, NK and T cells | Regulates NK, T, and DC activation state | Zhang et al. 2017 [91] Haubner et al. 2019 [24] Quek et al. 2016 [36] Agresta et al. 2018 [92] |
| GPR56 | N.D | No | Yes | Central nervous system, T cells | Frontal cortex development, NK inhibition, cell migration, HSC generation | Pabst et al. 2016 [93] Daga et al. 2019 [55] Kartalaei et al. 2015 [94] Huang et al. 2018 [95] |
| NKG2DL (its absence defines LSC) | Highly variable | Yes | No | Not expressed on healthy cells | Upregulation of NG2DL on malignant or virus-infected cells resulting in their clearance by NK cells | Paczulla et al. 2019 [6] Zingoni et al. 2018 [96] |
| TIM-3 | 98 | Yes | No | T cells, monocytes, macrophages, DC, and mast cells | Homeostasis-maintaining molecule of the immune system | Jan et al. 2011 [97] Haubner et al. 2019 [24] Kikushige et al. 2010 [98] Han et al. 2013 [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnone, M.; Konantz, M.; Hanns, P.; Paczulla Stanger, A.M.; Bertels, S.; Godavarthy, P.S.; Christopeit, M.; Lengerke, C. Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers 2020, 12, 3742. https://doi.org/10.3390/cancers12123742
Arnone M, Konantz M, Hanns P, Paczulla Stanger AM, Bertels S, Godavarthy PS, Christopeit M, Lengerke C. Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers. 2020; 12(12):3742. https://doi.org/10.3390/cancers12123742
Chicago/Turabian StyleArnone, Marlon, Martina Konantz, Pauline Hanns, Anna M. Paczulla Stanger, Sarah Bertels, Parimala Sonika Godavarthy, Maximilian Christopeit, and Claudia Lengerke. 2020. "Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity" Cancers 12, no. 12: 3742. https://doi.org/10.3390/cancers12123742
APA StyleArnone, M., Konantz, M., Hanns, P., Paczulla Stanger, A. M., Bertels, S., Godavarthy, P. S., Christopeit, M., & Lengerke, C. (2020). Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers, 12(12), 3742. https://doi.org/10.3390/cancers12123742