Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib

 ,
,  ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
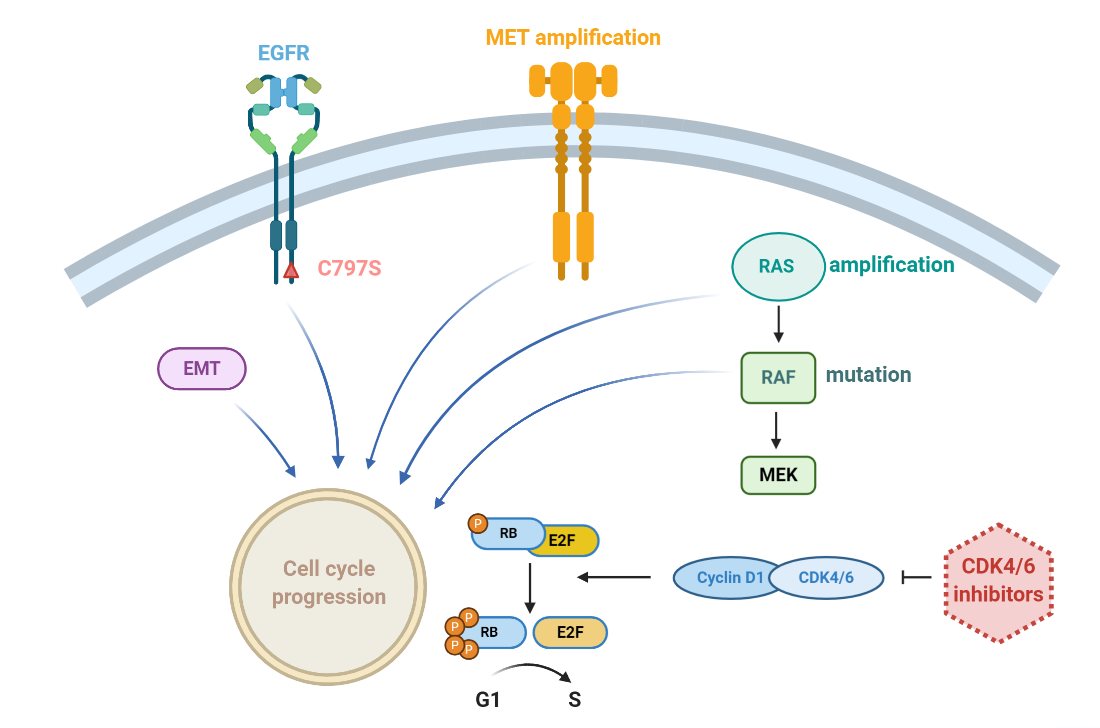
1. Introduction
2. Results
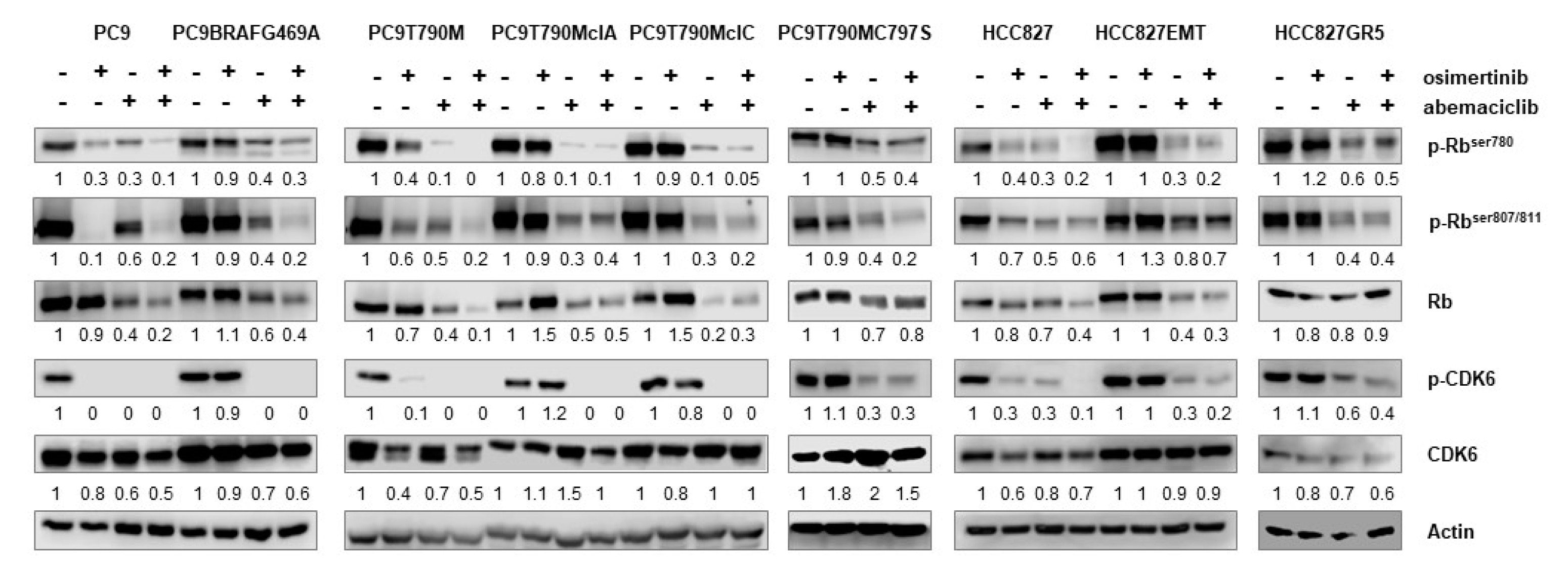
2.1. Osimertinib-Resistant Cell Models Up-Regulate the Phosphorylation of Rb Protein and Display Sensitivity to CDK4/6 Inhibition
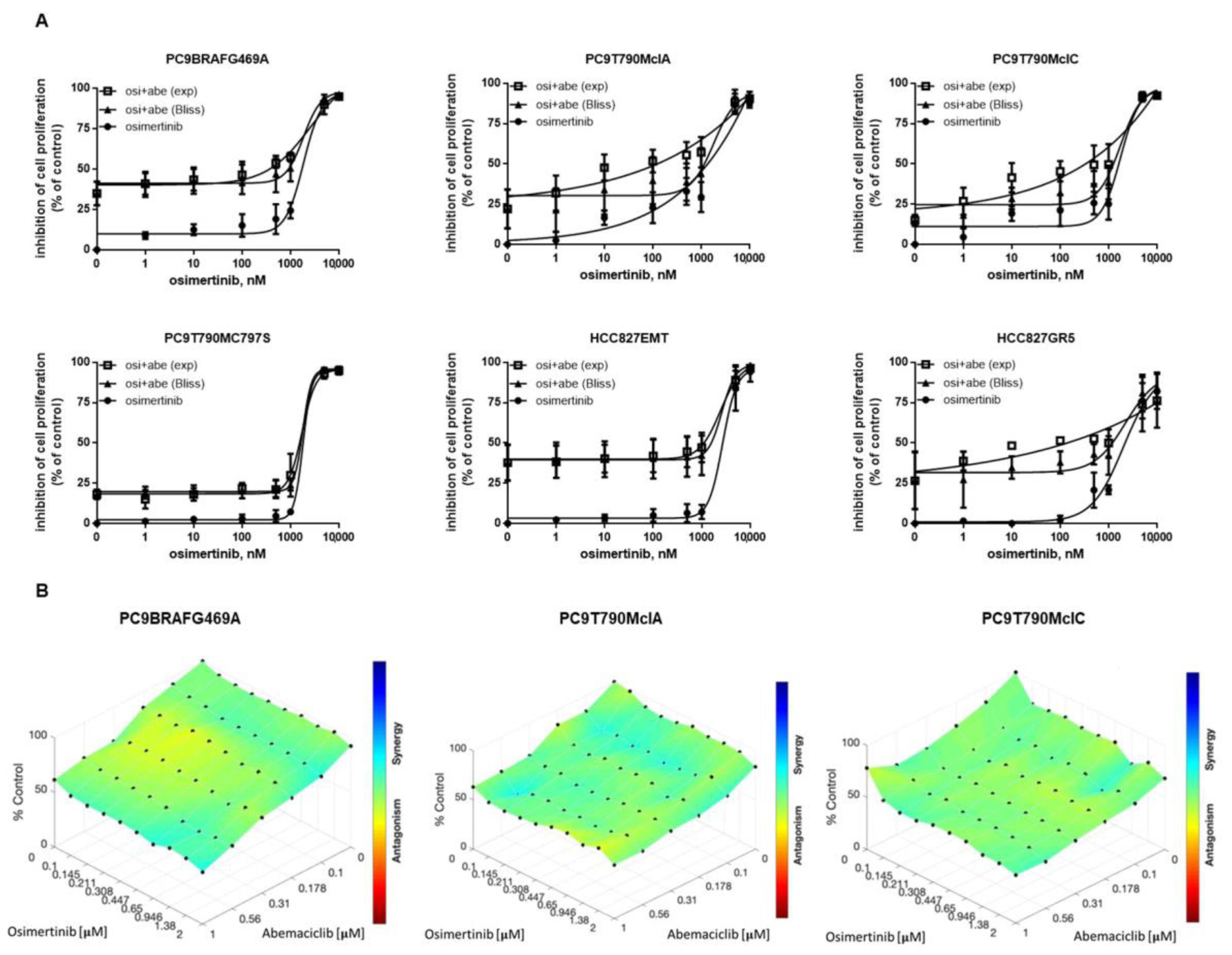
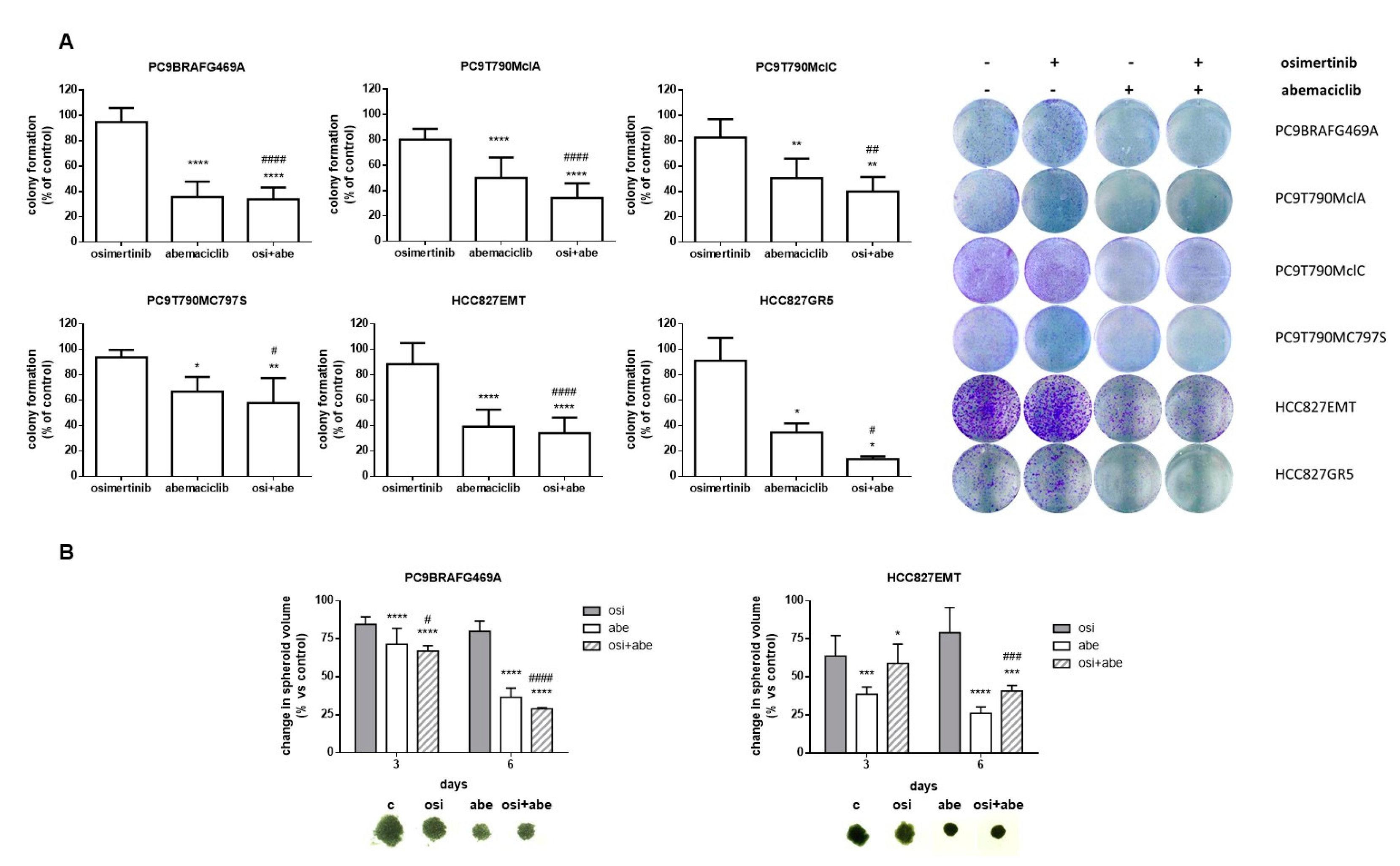
2.2. Effects of Abemaciclib Alone or Combined with Osimertinib on 2-D and 3-D Cell Growth and on the Expression of Cell Cycle-Related Proteins
2.3. Effects of Abemaciclib Alone or Combined with Osimertinib on Cell Death and Senescence
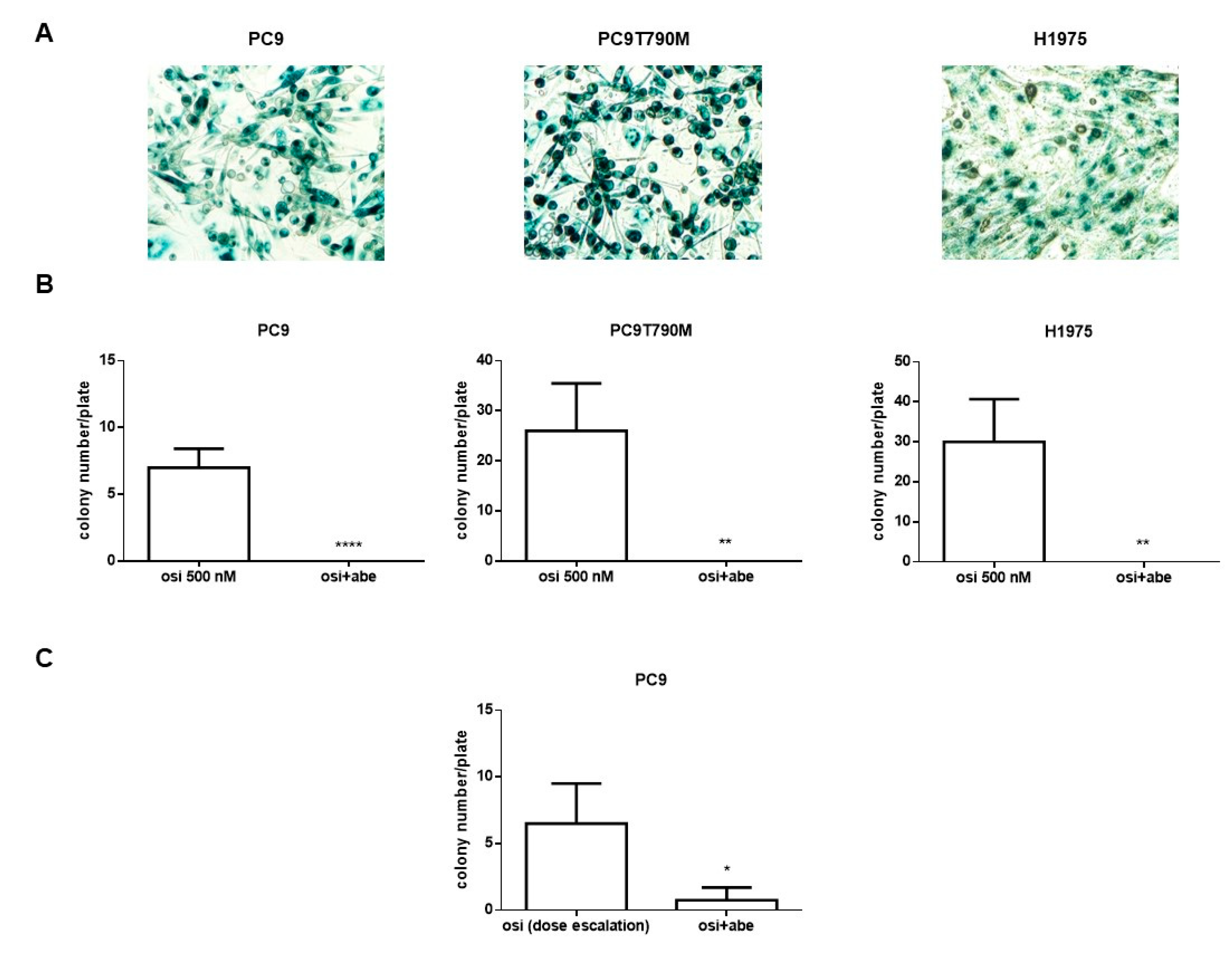
2.4. Abemaciclib Combined with Osimertinib Prevents the Appearance of Osimertinib Resistance
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Drug Treatment
4.3. Analysis of Cell Proliferation, Cell Death and Cell Cycle
4.4. Spheroid Generation
4.5. Colony Formation Assay
4.6. Senescence Evaluation
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Ahn, M.-J.; Tsai, C.; Shepherd, F.A.; Bazhenova, L.; Sequist, L.V.; Hida, T.; Yang, J.C.H.; Ramalingam, S.S.; Mitsudomi, T.; Jänne, P.A.; et al. Osimertinib in patients with T790M mutation-positive, advanced non–small cell lung cancer: Long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer 2019, 125, 892–901. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Gelsomino, F. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2− Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Bonelli, M.; La Monica, S.; Fumarola, C.; Alfieri, R. Multiple effects of CDK4/6 inhibition in cancer: From cell cycle arrest to immunomodulation. Biochem. Pharmacol. 2019, 170, 113676. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Zhou, X.; Shuzhang, D.; Nie, C.; Zhang, X.; Huang, J. Palbociclib overcomes afatinib resistance in non-small cell lung cancer. Biomed. Pharmacother. 2019, 109, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Comisar, C.; Witjes, H.; Maringwa, J.; De Greef, R.; Vishwanathan, K.; Cantarini, M.; Cox, E. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br. J. Clin. Pharmacol. 2017, 83, 1216–1226. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell. Chem. Biol. 2019, 26, 1067–1080. [Google Scholar] [CrossRef]
- Naz, S.; Sowers, A.; Choudhuri, R.; Wissler, M.; Gamson, J.; Mathias, A.; Cook, J.A.; Mitchell, J.B. Abemaciclib, a Selective CDK4/6 Inhibitor, Enhances the Radiosensitivity of Non-Small Cell Lung Cancer In Vitro and In Vivo. Clin. Cancer Res. 2018, 24, 3994–4005. [Google Scholar] [CrossRef]
- Prives, C.; White, E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008, 22, 1259–1264. [Google Scholar] [CrossRef]
- Wu, S.; Çetinkaya, C.; Muñoz-Alonso, M.J.; Von Der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; León, J.; Larsson, L.-G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef]
- Xu, X.; Lu, Z.; Qiang, W.; Vidimar, V.; Kong, B.; Kim, J.J.; Wei, J.-J. Inactivation of AKT Induces Cellular Senescence in Uterine Leiomyoma. Endocrinology 2014, 155, 1510–1519. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.J.F.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Leontieva, O.V.; Blagosklonny, M.V. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: Duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013, 12, 3063–3069. [Google Scholar] [CrossRef]
- Wu, C.-H.; Van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef] [PubMed]
- Freed, D.M.; Hall, C.R.; Strum, J.C.; Roberts, P.J. CDK4/6 inhibition with lerociclib (G1T38) delays acquired resistance to targeted therapies in preclinical models of non-small cell lung cancer. Cancer Res. 2019, 79 (Suppl. 13). [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.P.; A Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.B.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef]
- Bhateja, P.; Chiu, M.; Wildey, G.; Lipka, M.B.; Fu, P.; Yang, M.C.L.; Ardeshir-Larijani, F.; Sharma, N.; Dowlati, A. Retinoblastoma mutation predicts poor outcomes in advanced non small cell lung cancer. Cancer Med. 2019, 8, 1459–1466. [Google Scholar] [CrossRef]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; E McCoach, C.; McGranahan, N.; A Wilson, G.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Bulusu, K.C.; Lauset, G.; et al. LBA51 Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, VIII741. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. LBA50 Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, VIII740. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef] [PubMed]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Minari, R.; Cretella, D.; Flammini, L.; Fumarola, C.; Bonelli, M.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Madeddu, D.; et al. Third generation EGFR inhibitor osimertinib combined with pemetrexed or cisplatin exerts long-lasting anti-tumor effect in EGFR-mutated pre-clinical models of NSCLC. J. Exp. Clin. Cancer Res. 2019, 38, 222. [Google Scholar] [CrossRef] [PubMed]
- Rebuzzi, S.E.; Alfieri, R.; La Monica, S.; Minari, R.; Petronini, P.G.; Tiseo, M. Combination of EGFR-TKIs and chemotherapy in advanced EGFR mutated NSCLC: Review of the literature and future perspectives. Crit. Rev. Oncol. 2020, 146, 102820. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef]
- Yu, H.A.; Schoenfeld, A.J.; Makhnin, A.; Kim, R.; Rizvi, H.; Tsui, D.; Falcon, C.; Houck-Loomis, B.; Meng, F.; Yang, J.L.; et al. Effect of Osimertinib and Bevacizumab on Progression-Free Survival for Patients With Metastatic EGFR-Mutant Lung Cancers: A Phase 1/2 Single-Group Open-Label Trial. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.-B.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- La Monica, S.; Madeddu, D.; Tiseo, M.; Vivo, V.; Galetti, M.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Falco, A.; et al. Combination of Gefitinib and Pemetrexed Prevents the Acquisition of TKI Resistance in NSCLC Cell Lines Carrying EGFR- Activating Mutation. J. Thorac. Oncol. 2016, 11, 1051–1063. [Google Scholar] [CrossRef]
- Meder, L.; König, K.; Ozretić, L.; Schultheis, A.M.; Ueckeroth, F.; Ade, C.P.; Albus, K.; Boehm, D.; Rommerscheidt-Fuss, U.; Florin, A.; et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int. J. Cancer 2015, 138, 927–938. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Chou, C.-W.; Lin, C.-H.; Hsiao, T.-H.; Lo, C.-C.; Hsieh, C.-Y.; Huang, C.-C.; Sher, Y.-P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Minari, R.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Galetti, M.; Digiacomo, G.; Riccardi, F.; Petronini, P.G.; et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Target. Oncol. 2019, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Gal, H.; Gaborit, N.; Mazzeo, L.; Romaniello, D.; Salame, T.M.; Lindzen, M.; Mahlknecht, G.; Enuka, Y.; Burton, D.G.; et al. An oligoclonal antibody durably overcomes resistance of lung cancer to third-generation EGFR inhibitors. EMBO Mol. Med. 2017, 10, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Cavazzoni, A.; Alfieri, R.R.; Carmi, C.; Zuliani, V.; Galetti, M.; Fumarola, C.; Frazzi, R.; Bonelli, M.; Bordi, F.; Lodola, A.; et al. Dual mechanisms of action of the 5-benzylidene-hydantoin UPR1024 on lung cancer cell lines. Mol. Cancer Ther. 2008, 7, 361–370. [Google Scholar] [CrossRef]
- Fumarola, C.; La Monica, S.; Alfieri, R.R.; Borra, E.; Guidotti, G.G. Cell size reduction induced by inhibition of the mTOR/S6K-signaling pathway protects Jurkat cells from apoptosis. Cell Death Differ. 2005, 12, 1344–1357. [Google Scholar] [CrossRef]
- La Monica, S.; Galetti, M.; Alfieri, R.; Cavazzoni, A.; Ardizzoni, A.; Tiseo, M.; Capelletti, M.; Goldoni, M.; Tagliaferri, S.; Mutti, A.; et al. Everolimus restores gefitinib sensitivity in resistant non-small cell lung cancer cell lines. Biochem. Pharmacol. 2009, 78, 460–468. [Google Scholar] [CrossRef]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef]
- La Monica, S.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Digiacomo, G.; Flammini, L.; Barocelli, E.; Minari, R.; Naldi, N.; et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 1–12. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers 2021, 13, 6. https://doi.org/10.3390/cancers13010006
La Monica S, Fumarola C, Cretella D, Bonelli M, Minari R, Cavazzoni A, Digiacomo G, Galetti M, Volta F, Mancini M, et al. Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers. 2021; 13(1):6. https://doi.org/10.3390/cancers13010006
Chicago/Turabian StyleLa Monica, Silvia, Claudia Fumarola, Daniele Cretella, Mara Bonelli, Roberta Minari, Andrea Cavazzoni, Graziana Digiacomo, Maricla Galetti, Francesco Volta, Maicol Mancini, and et al. 2021. "Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib" Cancers 13, no. 1: 6. https://doi.org/10.3390/cancers13010006
APA StyleLa Monica, S., Fumarola, C., Cretella, D., Bonelli, M., Minari, R., Cavazzoni, A., Digiacomo, G., Galetti, M., Volta, F., Mancini, M., Petronini, P. G., Tiseo, M., & Alfieri, R. (2021). Efficacy of the CDK4/6 Dual Inhibitor Abemaciclib in EGFR-Mutated NSCLC Cell Lines with Different Resistance Mechanisms to Osimertinib. Cancers, 13(1), 6. https://doi.org/10.3390/cancers13010006