Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
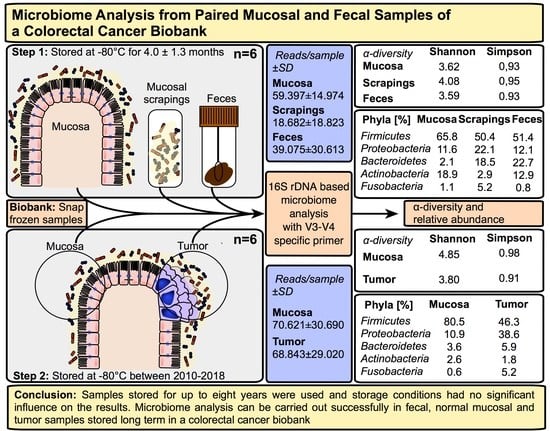
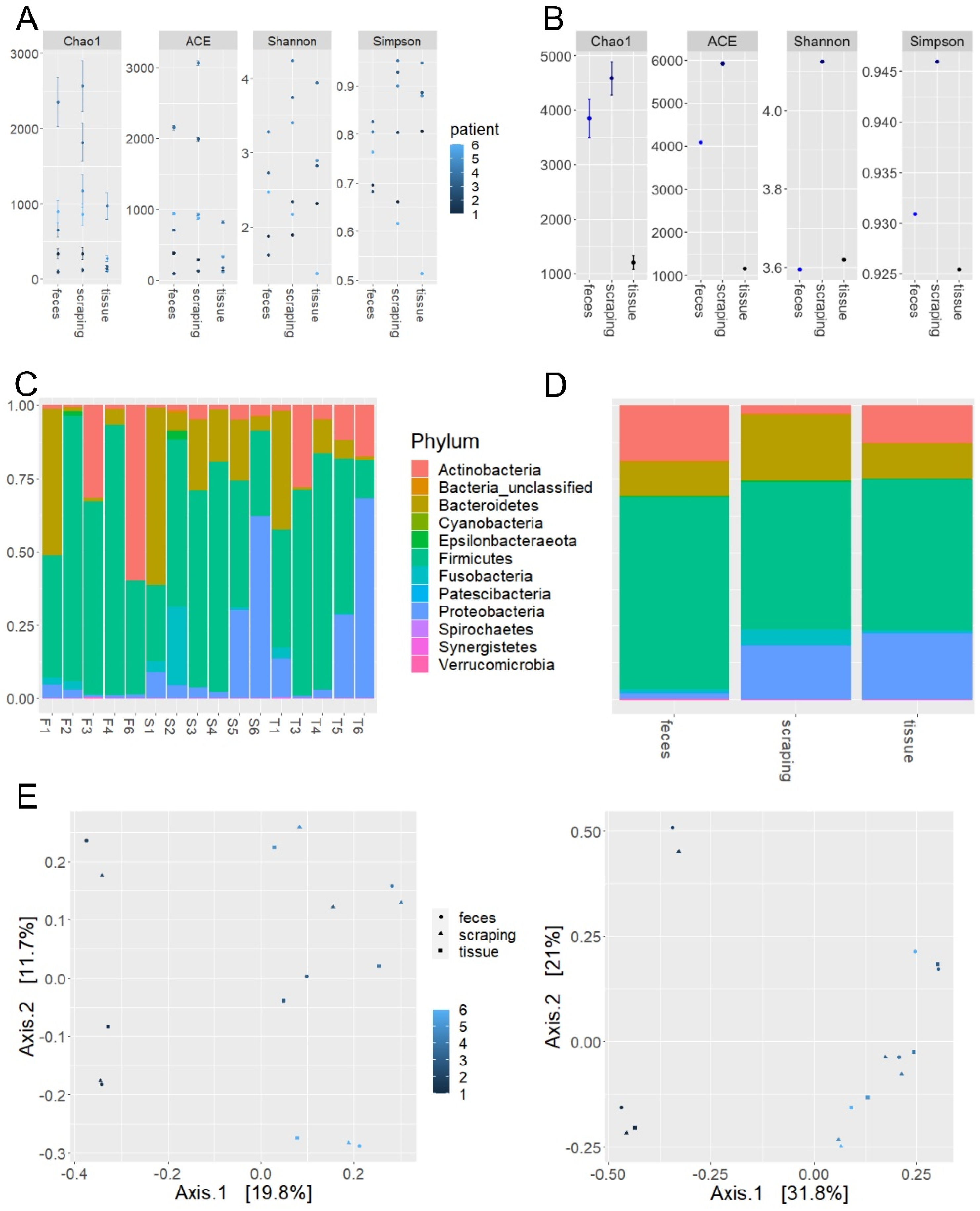
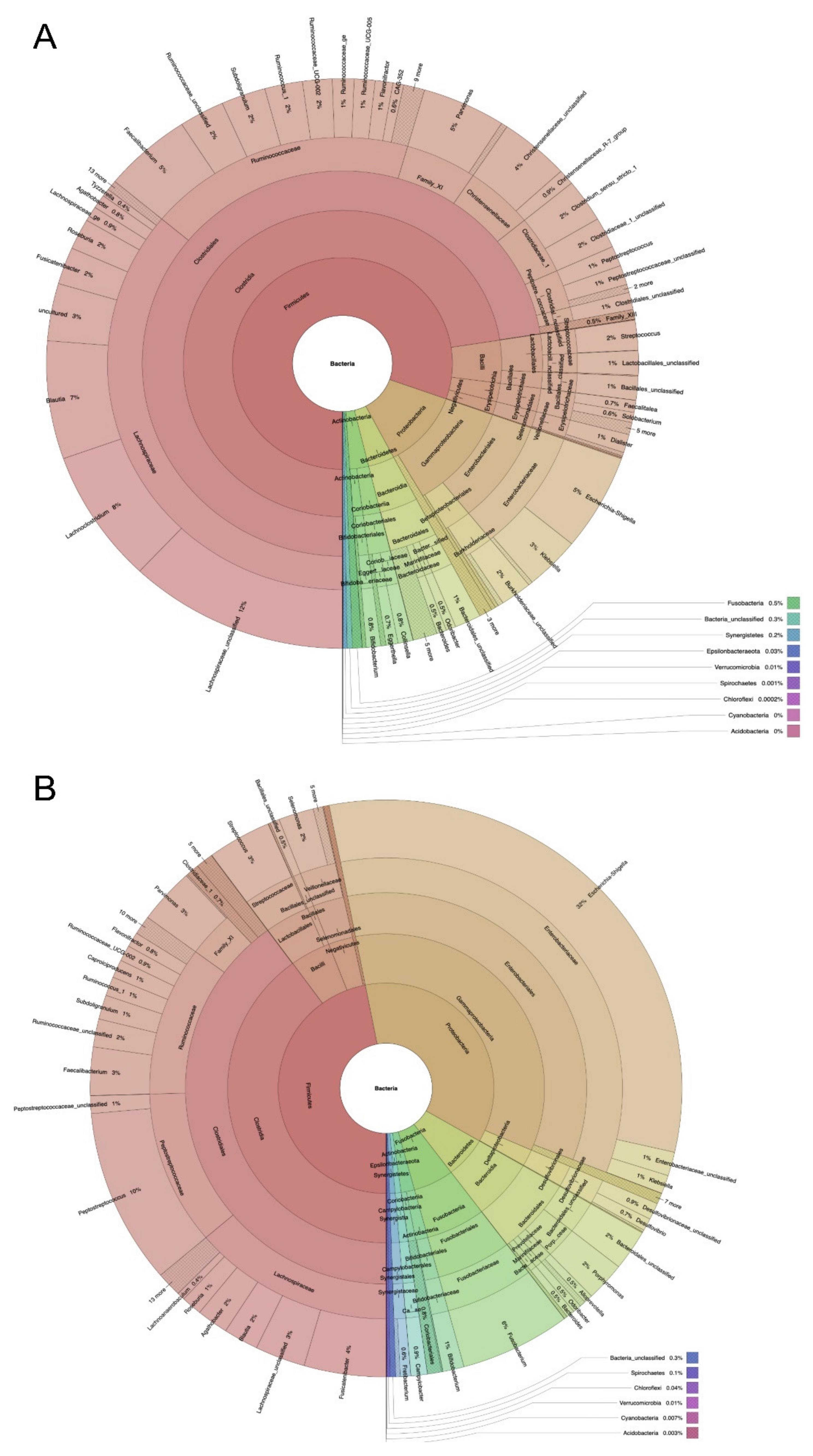
2.1. Microbiome Profiling of Luminal and Mucosal Sample Sites (Step 1)
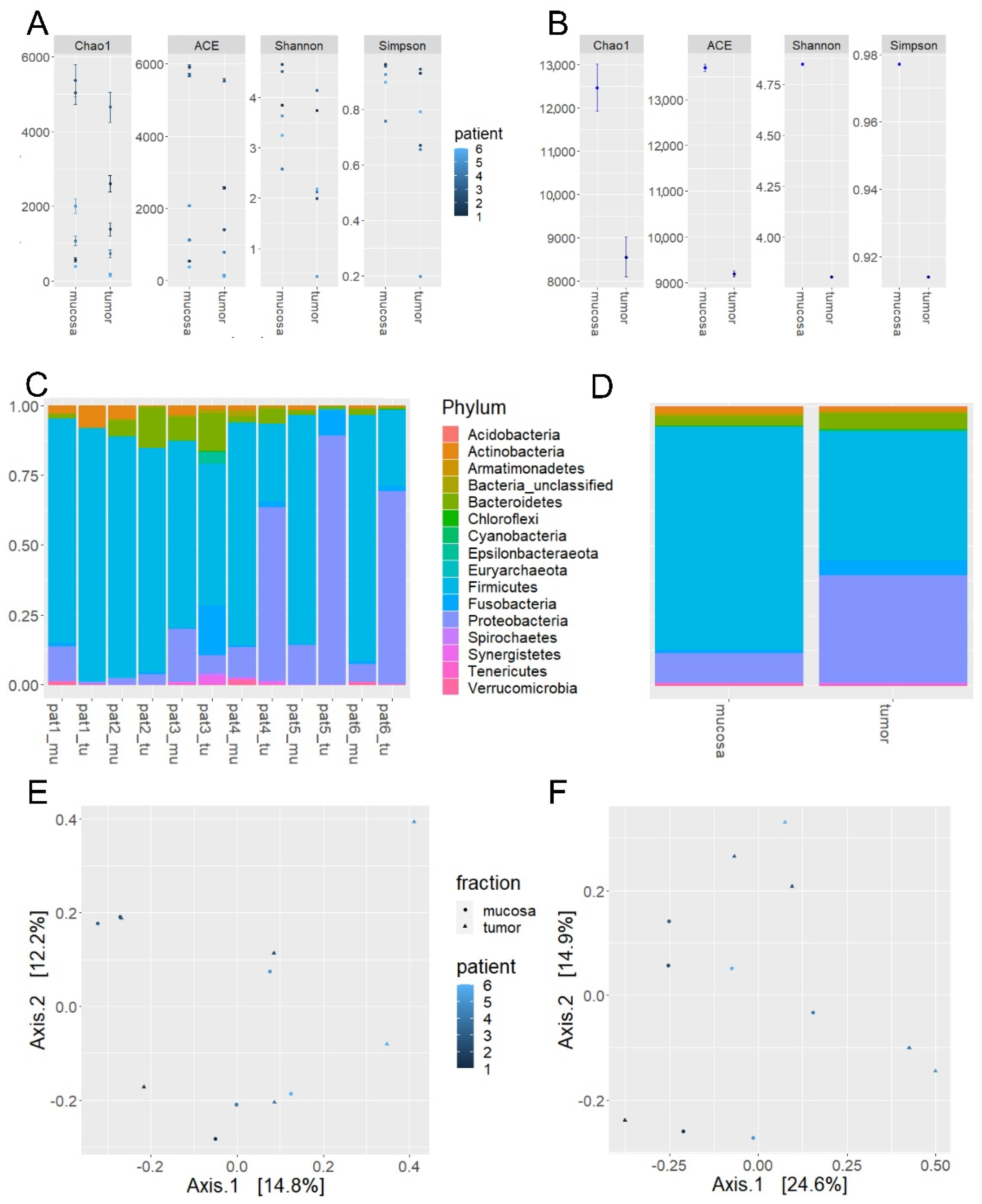
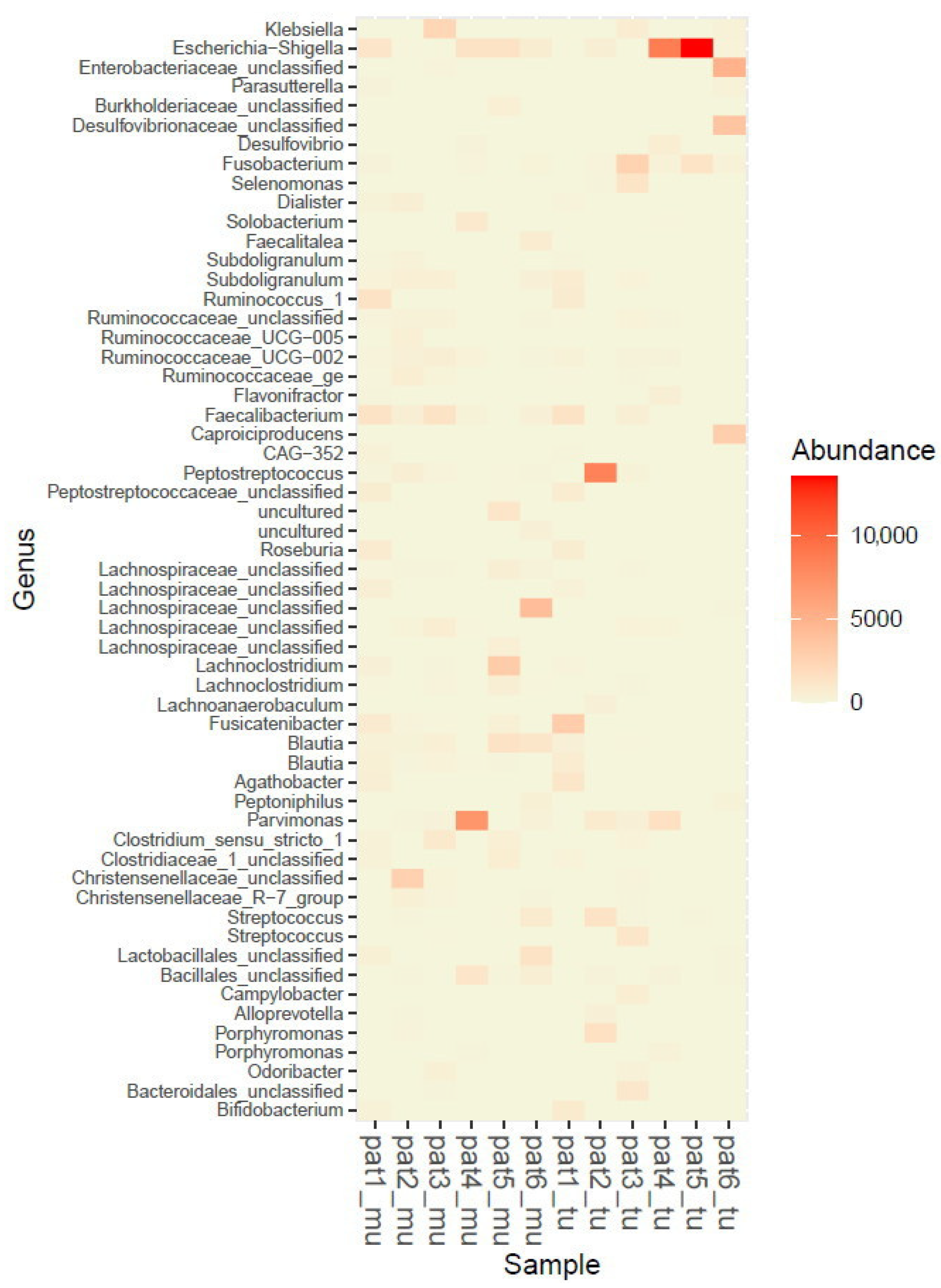
2.2. Microbiome Profiling from Tumor and Adjacent Normal Mucosa Samples as Well as Influence of Storage Duration (Step 2)
3. Discussion
4. Materials and Methods
4.1. Ethical Framework
4.2. Study Design and Sample Collection
4.3. DNA Extraction, Amplification and Sequencing
4.4. Bioinformatics and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, J.; Sears, C.L. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: Contributions to colorectal cancer development. Genome Med. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nat. Cell Biol. 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef]
- Barrington, W.T.; Lusis, A.J. Atherosclerosis: Association between the gut microbiome and atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 699–700. [Google Scholar] [CrossRef]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef]
- Belizário, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef]
- Gaines, S.; Shao, C.; Hyman, N.; Alverdy, J.C. Gut microbiome influences on anastomotic leak and recurrence rates following colorectal cancer surgery. BJS 2018, 105, e131–e141. [Google Scholar] [CrossRef]
- Mizutani, S.; Yamada, T.; Yachida, S. Significance of the gut microbiome in multistep colorectal carcinogenesis. Cancer Sci. 2020, 111, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Gethings-Behncke, C.; Coleman, H.G.; Jordao, H.W.; Longley, D.B.; Crawford, N.; Murray, L.J.; Kunzmann, A.T. Fusobacterium nucleatum in the Colorectum and Its Association with Cancer Risk and Survival: A Systematic Review and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2020, 29, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Klimesova, K.; Zakostelska, Z.J.; Tlaskalova-Hogenova, H. Oral Bacterial and Fungal Microbiome Impacts Colorectal Carcinogenesis. Front. Microbiol. 2018, 9, 774. [Google Scholar] [CrossRef]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- He, Y.; Wu, W.; Zheng, H.-M.; Li, P.; McDonald, D.; Sheng, H.-F.; Chen, M.-X.; Chen, Z.-H.; Ji, G.-Y.; Zheng, Z.-D.-X.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Koliarakis, I.; Athanasakis, E.; Sgantzos, M.; Mariolis-Sapsakos, T.; Xynos, E.; Chrysos, E.; Souglakos, J.; Tsiaoussis, J. Intestinal Microbiota in Colorectal Cancer Surgery. Cancers 2020, 12, 3011. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Gu, J. Clinical Implications of the Associations Between Intestinal Microbiome and Colorectal Cancer Progression. Cancer Manag. Res. 2020, 12, 4117–4128. [Google Scholar] [CrossRef]
- Drewes, J.L.; Housseau, F.; Sears, C.L. Sporadic colorectal cancer: Microbial contributors to disease prevention, development and therapy. Br. J. Cancer 2016, 115, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Vogtmann, E.; Ahlquist, D.A.; Devens, M.E.; Kisiel, J.B.; Taylor, W.R.; White, B.A.; Hale, V.L.; Sung, J.; Chia, N.; et al. Fecal Metabolomic Signatures in Colorectal Adenoma Patients Are Associated with Gut Microbiota and Early Events of Colorectal Cancer Pathogenesis. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Jiang, B. Analysis of Mucosa-Associated Microbiota in Colorectal Cancer. Med. Sci. Monit. 2017, 23, 4422–4430. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Kim, N.; Park, J.H.; Kim, Y.S.; Lee, J.; Kim, H.W.; Choi, Y.J.; Shin, C.M.; Park, Y.S.; Lee, N.H.; et al. Comparisons of Gut Microbiota Among Healthy Control, Patients with Conventional Adenoma, Sessile Serrated Adenoma, and Colorectal Cancer. J. Cancer Prev. 2017, 22, 108–114. [Google Scholar] [CrossRef]
- Alverdy, J.C.; Hyoju, S.K.; Weigerinck, M.; Gilbert, J.A. The gut microbiome and the mechanism of surgical infection. BJS 2017, 104, e14–e23. [Google Scholar] [CrossRef]
- Guyton, K.; Alverdy, J.C. The gut microbiota and gastrointestinal surgery. Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Vasapolli, R.; Schütte, K.; Schulz, C.; Vital, M.; Schomburg, D.; Pieper, D.H.; Vilchez-Vargas, R.; Malfertheiner, P. Analysis of Transcriptionally Active Bacteria throughout the Gastrointestinal Tract of Healthy Individuals. Gastroenterology 2019, 157, 1081–1092.e3. [Google Scholar] [CrossRef]
- Codling, C.; O’Mahony, L.; Shanahan, F.; Quigley, E.M.M.; Marchesi, J.R. A Molecular Analysis of Fecal and Mucosal Bacterial Communities in Irritable Bowel Syndrome. Dig. Dis. Sci. 2009, 55, 392–397. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Chen, J.; Chen, X.; Chia, N.; O’Connor, H.M.; Wolf, P.G.; Gaskins, H.R.; Bharucha, A.E. Relationship between Microbiota of the Colonic Mucosa vs. Feces and Symptoms, Colonic Transit, and Methane Production in Female Patients with Chronic Constipation. Gastroenterology 2016, 150, 367–379.e1. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gomez-Millan, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Genet. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.L.; Schelcher, C.; Gashi, S.; Schreiber, S.; Thasler, R.M.K.; Jauch, K.-W.; Thasler, W.E. RNA Stability in Human Liver: Comparison of Different Processing Times, Temperatures and Methods. Mol. Biotechnol. 2013, 53, 1–8. [Google Scholar] [CrossRef]
- Lee, S.M.L.; Schelcher, C.; Thasler, R.; Schiergens, T.S.; Thasler, W.E. Pre-Analytical Determination of the Effect of Extended Warm or Cold Ischaemia on RNA Stability in the Human Ileum Mucosa. PLoS ONE 2015, 10, e0138214. [Google Scholar] [CrossRef] [PubMed]
- Thasler, W.E.; Weiss, T.S.; Schillhorn, K.; Stoll, P.-T.; Irrgang, B.; Jauch, K.-W. Charitable State-Controlled Foundation Human Tissue and Cell Research: Ethic and Legal Aspects in the Supply of Surgically Removed Human Tissue for Research in the Academic and Commercial Sector in Germany. Cell Tissue Bank. 2003, 4, 49–56. [Google Scholar] [CrossRef]
- Thasler, W.E.; Thasler, R.M.K.; Schelcher, C.; Jauch, K.-W. Biobanking for research in surgery: Are surgeons in charge for advancing translational research or mere assistants in biomaterial and data preservation? Langenbeck’s Arch. Surg. 2013, 398, 487–499. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Luan, Y.; Sun, F. Variance adjusted weighted UniFrac: A powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinform. 2011, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. vegan: Community Ecology Package R Package Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 12 October 2020).
- Wickham, H. Ggplot2. Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Collection Site | Weight (g) of Samples (Mean) | IT 1 (min) OR 2 until Sampling (Mean ± SD) | IT 1 (min) Sampling until Freezing (Mean ± SD) | Duration (Months) of Storage at −80 °C (Mean ± SD) | |
|---|---|---|---|---|---|
| Step 1 n = 6 | Feces | 0.19 | 36.3 ± 23.8 | 114.3 ± 95.6 | 4.0 ± 1.3 |
| Mucosa | 0.23 | ||||
| Scraping | 0.36 | ||||
| Step 2 n = 6 | Mucosa | 0.20 | 39.2 ± 20.3 | 37.8 ± 10.0 | 59.0 ± 37.6 |
| Tumor | 0.26 |
| Basic Data | α-Diversity Measures | |||||||
|---|---|---|---|---|---|---|---|---|
| gDNA (ng/µL) (PicoGreen) | DNA Concentration (ng/µL) after Amplification | Reads/Sample | Chao1 | ACE | Shannon | Simpson | ||
| Feces | 1 | 103.88 | 9.95 | 38,830 | 3848 | 4094 | 3.59 | 0.93 |
| 2 | 186.72 | 7.98 | 61,629 | |||||
| 3 | 143.18 | 4.23 | 4733 | |||||
| 4 | 163.34 | 3.26 | 54,584 | |||||
| 5 | 165.46 | 0.04 | 249 * | |||||
| 6 | 116.20 | 6.32 | 74,427 | |||||
| Mean ± SD | 146.5 ± 31.6 | 5.30 ± 3.50 | 39,075 ± 30,613 | |||||
| Mucosa | 1 | 182.15 | 3.17 | 46,077 | 1207 | 1163 | 3.62 | 0.93 |
| 2 | 172.71 | 0.27 | 173 * | |||||
| 3 | 174.29 | 3.60 | 6058 | |||||
| 4 | 167.36 | 5.82 | 8294 | |||||
| 5 | 179.41 | 7.84 | 13,265 | |||||
| 6 | 171.14 | 5.32 | 38,225 | |||||
| Mean ± SD | 174.51 ± 5.45 | 4.34 ± 2.60 | 59,397 ± 14,974 | |||||
| Scraping | 1 | 18.95 | 7.25 | 62,021 | 6165 | 6064 | 4.08 | 0.95 |
| 2 | 42.91 | 1.68 | 45,152 | |||||
| 3 | 8.14 | 6.65 | 50,863 | |||||
| 4 | 118.70 | 8.98 | 84,086 | |||||
| 5 | 6.86 | 10.18 | 46,675 | |||||
| 6 | 6.04 | 4.33 | 67,589 | |||||
| Mean ± SD | 33.60 ± 43.97 | 6.51 ± 3.11 | 18,682 ± 18,823 | |||||
| Species | Feces | Scrapings | Mucosa |
|---|---|---|---|
| Firmicutes | 65.8 | 50.4 | 51.4 |
| Bacteroidetes | 11.6 | 22.1 | 12.1 |
| Proteobacteria | 2.1 | 18.5 | 22.7 |
| Actinobacteria | 18.9 | 2.9 | 12.9 |
| Fusobacteria | 1.1 | 5.2 | 0.8 |
| Mucosa | Tumor | ||
|---|---|---|---|
| Reads/sample ± SD | 70,621 ± 30,690 | 68,843 ± 29,020 | |
| α-diversity measures | Chao1 | 12,465 | 8553 |
| ACE | 13,690 | 9191 | |
| Shannon | 4.85 | 3.80 | |
| Simpson | 0.98 | 0.91 | |
| Relative abundance | Firmicutes | 80.5 | 46.3 |
| Proteobacteria | 10.9 | 38.6 | |
| Bacteroidetes | 3.6 | 5.9 | |
| Actinobacteria | 2.6 | 1.8 | |
| Fusobacteria | 0.6 | 5.2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wirth, U.; Garzetti, D.; Jochum, L.M.; Spriewald, S.; Kühn, F.; Ilmer, M.; Lee, S.M.L.; Niess, H.; Bazhin, A.V.; Andrassy, J.; et al. Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank. Cancers 2020, 12, 3702. https://doi.org/10.3390/cancers12123702
Wirth U, Garzetti D, Jochum LM, Spriewald S, Kühn F, Ilmer M, Lee SML, Niess H, Bazhin AV, Andrassy J, et al. Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank. Cancers. 2020; 12(12):3702. https://doi.org/10.3390/cancers12123702
Chicago/Turabian StyleWirth, Ulrich, Debora Garzetti, Lara M. Jochum, Stefanie Spriewald, Florian Kühn, Matthias Ilmer, Serene M. L. Lee, Hanno Niess, Alexandr V. Bazhin, Joachim Andrassy, and et al. 2020. "Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank" Cancers 12, no. 12: 3702. https://doi.org/10.3390/cancers12123702
APA StyleWirth, U., Garzetti, D., Jochum, L. M., Spriewald, S., Kühn, F., Ilmer, M., Lee, S. M. L., Niess, H., Bazhin, A. V., Andrassy, J., Werner, J., Stecher, B., & Schiergens, T. S. (2020). Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank. Cancers, 12(12), 3702. https://doi.org/10.3390/cancers12123702