Dysbiosis Triggers ACF Development in Genetically Predisposed Subjects

 , ,
, ,  ,
,  , , , , ,
, , , , ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
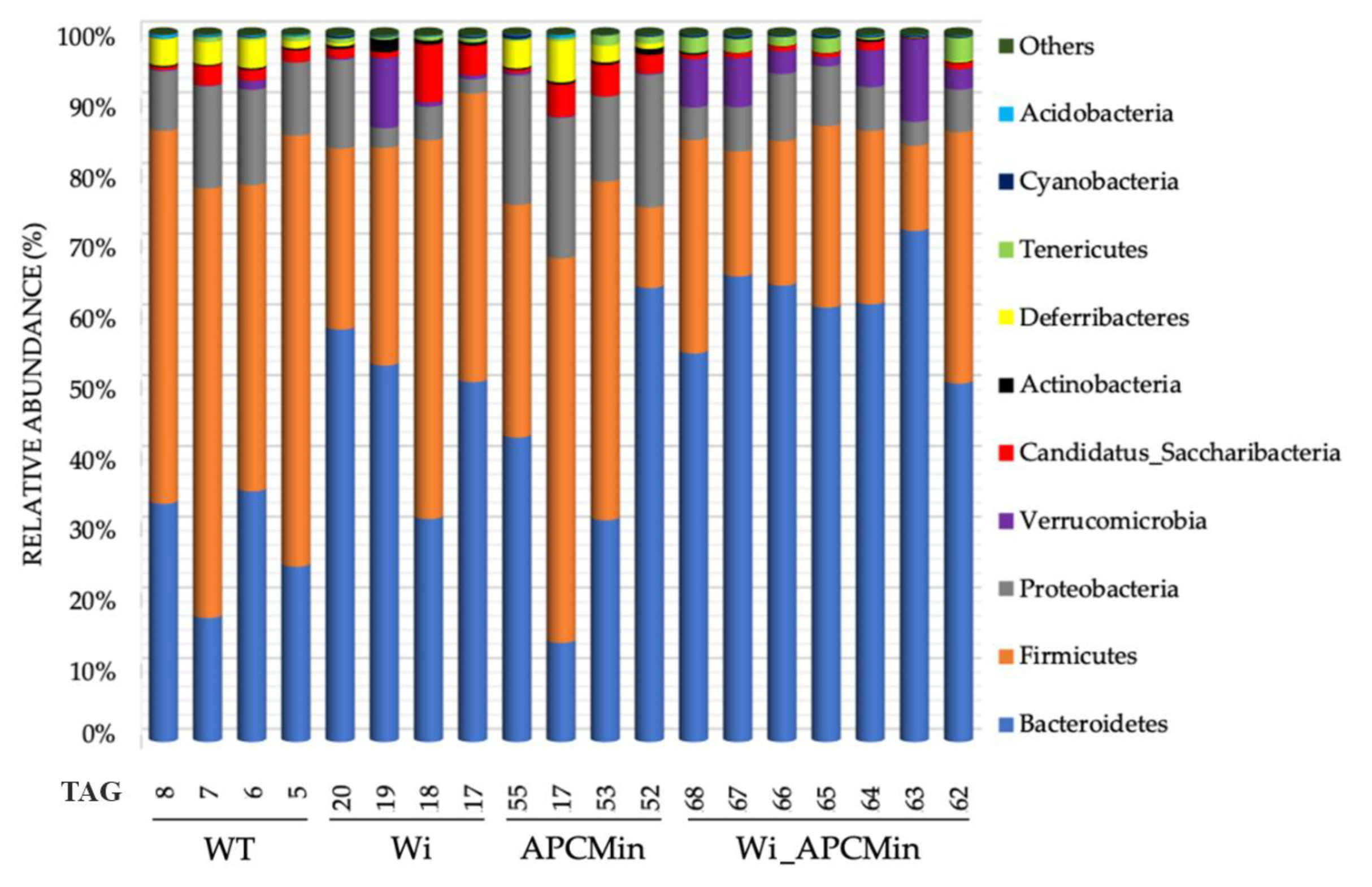
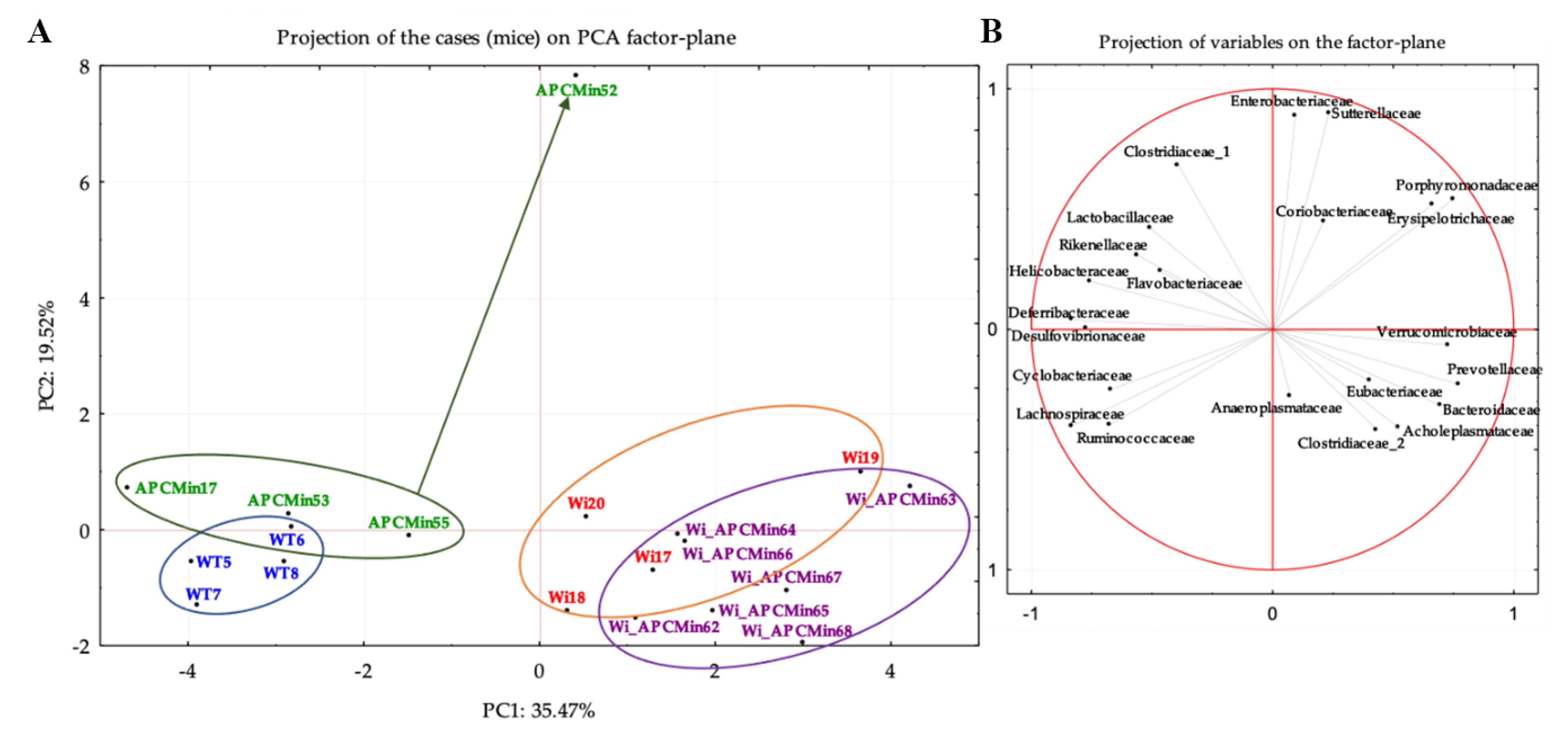
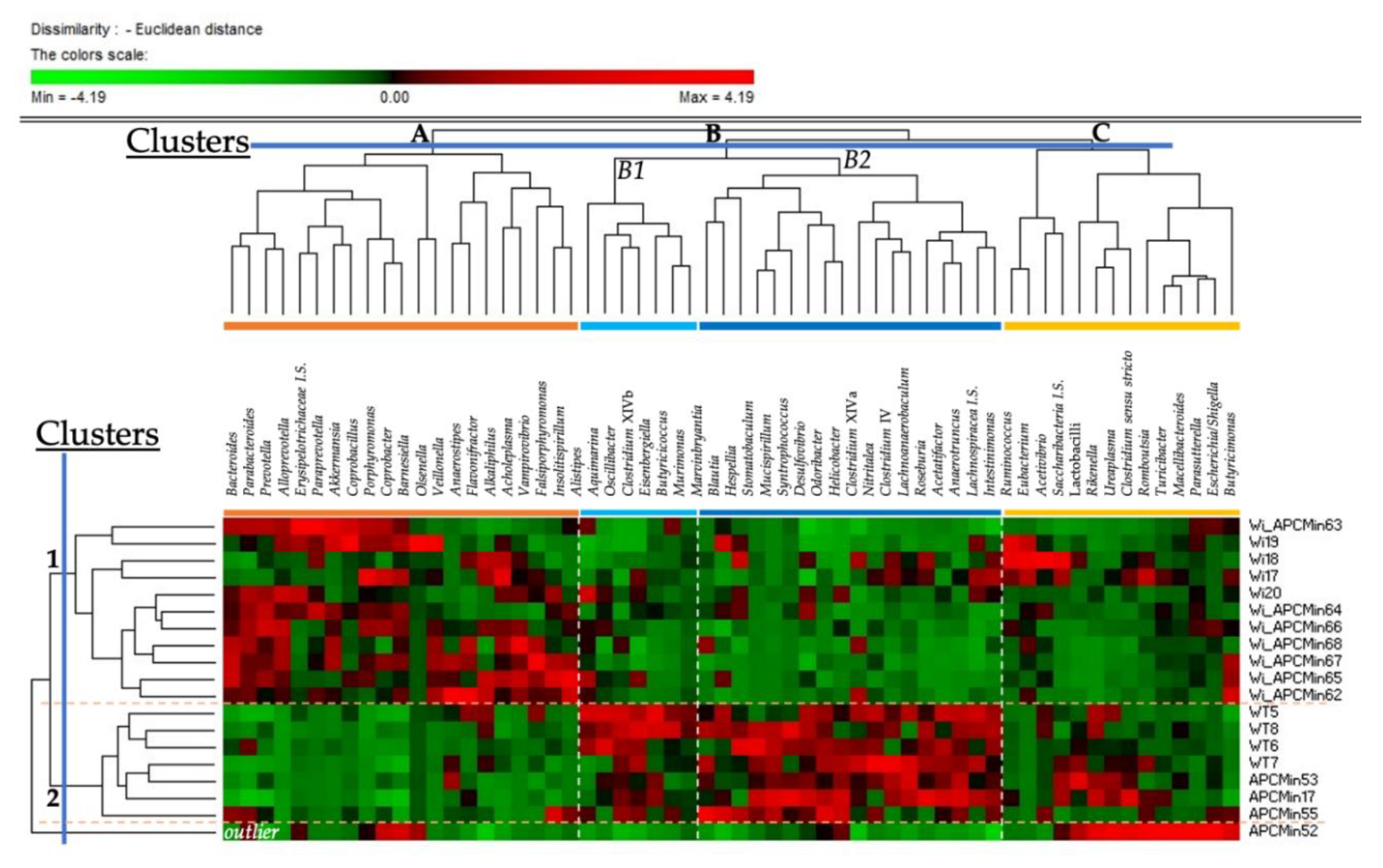
2.1. Intestinal Microbiota in Winnie-APCMin/+ Mice Shows Dysbiotic Features Typical of Winnie Mice
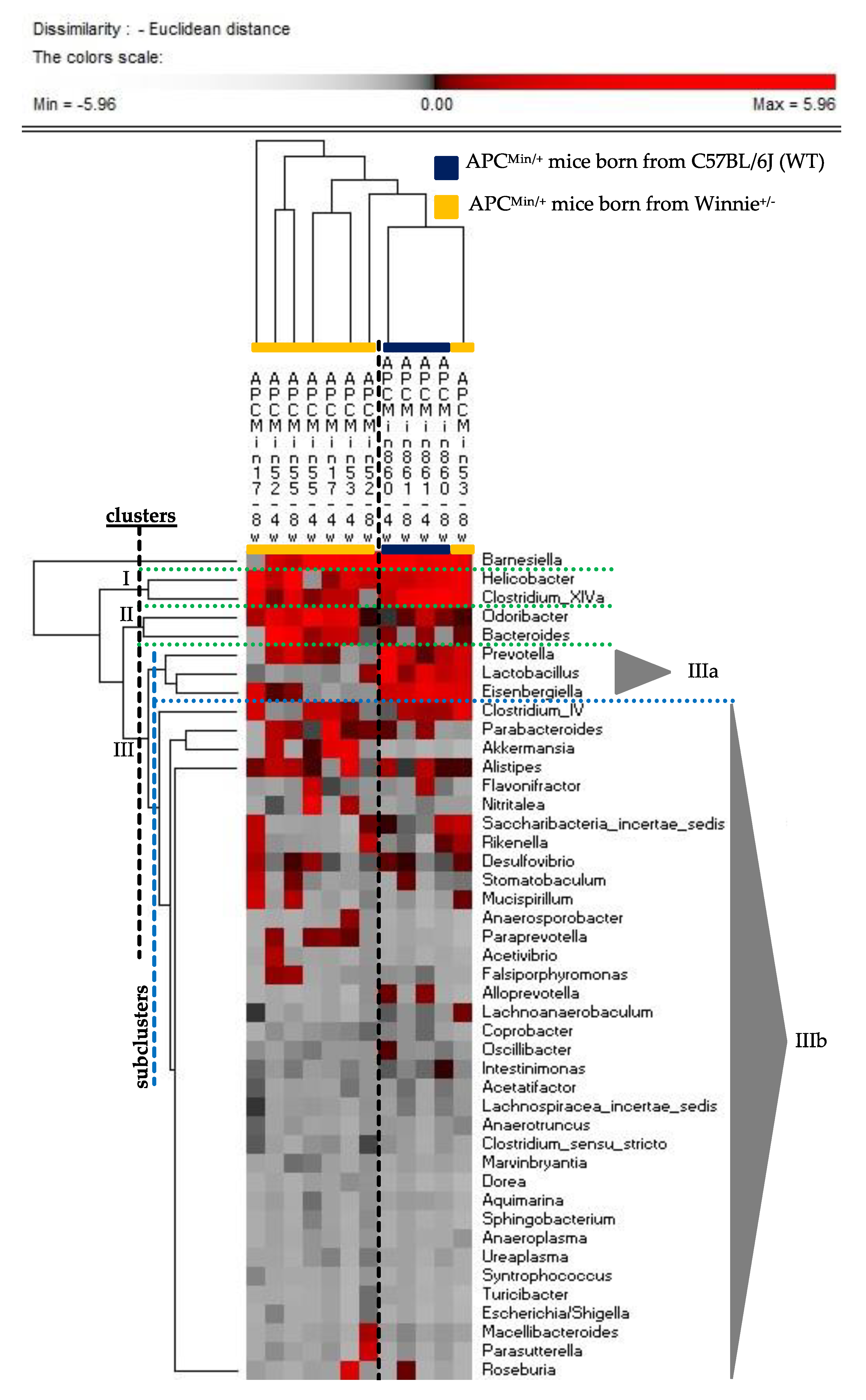
2.2. The Genetics of Breeding Couple Shapes APCMin/+ Mice Intestinal Microbiota
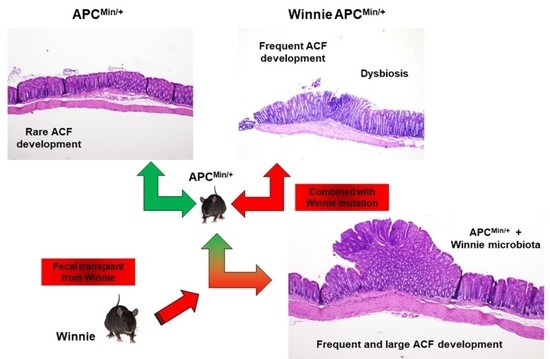
2.3. Fecal Transplant from Winnie to APCMin/+ Mice Triggers ACF Development
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Histology
4.3. DNA Extraction from Stool
4.4. S rDNA Metagenomic Library Preparation, Sequencing and Analysis
4.5. Fecal Transplantation
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- De Santis, S.; Galleggiante, V.; Scandiffio, L.; Liso, M.; Sommella, E.; Sobolewski, A.; Spilotro, V.; Pinto, A.; Campiglia, P.; Serino, G.; et al. Secretory Leukoprotease Inhibitor (Slpi) Expression Is Required for Educating Murine Dendritic Cells Inflammatory Response Following Quercetin Exposure. Nutrients 2017, 9, 706. [Google Scholar] [CrossRef]
- Cariello, M.; Contursi, A.; Gadaleta, R.M.; Piccinin, E.; De Santis, S.; Piglionica, M.; Spaziante, A.F.; Sabba, C.; Villani, G.; Moschetta, A. Extra-Virgin Olive Oil from Apulian Cultivars and Intestinal Inflammation. Nutrients 2020, 12, 1084. [Google Scholar] [CrossRef]
- De Santis, S.; Clodoveo, M.L.; Cariello, M.; D’Amato, G.; Franchini, C.; Faienza, M.F.; Corbo, F. Polyphenols and obesity prevention: Critical insights on molecular regulation, bioavailability and dose in preclinical and clinical settings. Crit. Rev. Food Sci. Nutr. 2020, 1–23. [Google Scholar] [CrossRef]
- Piccinin, E.; Cariello, M.; De Santis, S.; Ducheix, S.; Sabba, C.; Ntambi, J.M.; Moschetta, A. Role of Oleic Acid in the Gut-Liver Axis: From Diet to the Regulation of Its Synthesis via Stearoyl-CoA Desaturase 1 (SCD1). Nutrients 2019, 11, 2283. [Google Scholar] [CrossRef]
- De Santis, S.; Cariello, M.; Piccinin, E.; Sabba, C.; Moschetta, A. Extra Virgin Olive Oil: Lesson from Nutrigenomics. Nutrients 2019, 11, 2085. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Elinav, E. Diet-microbiota interactions and personalized nutrition. Nat. Rev. Microbiol. 2019, 17, 742–753. [Google Scholar] [CrossRef]
- Baquero, F.; Nombela, C. The microbiome as a human organ. Clin. Microbiol. Infect. 2012, 18, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining Dysbiosis for a Cluster of Chronic Diseases. Sci. Rep. 2019, 9, 12918. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Arques, A.; Scharl, M. Intestinal microbiota and colorectal carcinoma: Implications for pathogenesis, diagnosis, and therapy. EBioMedicine 2019, 48, 648–655. [Google Scholar] [CrossRef]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef]
- Wu, S.; Morin, P.J.; Maouyo, D.; Sears, C.L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 2003, 124, 392–400. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef]
- Gagniere, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef]
- Vogtmann, E.; Hua, X.; Zeller, G.; Sunagawa, S.; Voigt, A.Y.; Hercog, R.; Goedert, J.J.; Shi, J.; Bork, P.; Sinha, R. Colorectal Cancer and the Human Gut Microbiome: Reproducibility with Whole-Genome Shotgun Sequencing. PLoS ONE 2016, 11, e0155362. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Dechelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Eiseman, B.; Silen, W.; Bascom, G.S.; Kauvar, A.J. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 1958, 44, 854–859. [Google Scholar]
- Saus, E.; Iraola-Guzman, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldon, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Li, L.; Li, X.; Zhong, W.; Yang, M.; Xu, M.; Sun, Y.; Ma, J.; Liu, T.; Song, X.; Dong, W.; et al. Gut microbiota from colorectal cancer patients enhances the progression of intestinal adenoma in Apc(min/+) mice. EBioMedicine 2019, 48, 301–315. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Zhan, Y.; Chen, P.J.; Sadler, W.D.; Wang, F.; Poe, S.; Nunez, G.; Eaton, K.A.; Chen, G.Y. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 2013, 73, 7199–7210. [Google Scholar] [CrossRef]
- De Santis, S.; Verna, G.; Serino, G.; Armentano, R.; Cavalcanti, E.; Liso, M.; Dicarlo, M.; Coletta, S.; Mastronardi, M.; Lippolis, A.; et al. Winnie-APC(Min/+) Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation. Int. J. Mol. Sci. 2020, 21, 2972. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [PubMed]
- Liso, M.; De Santis, S.; Verna, G.; Dicarlo, M.; Calasso, M.; Santino, A.; Gigante, I.; Eri, R.; Raveenthiraraj, S.; Sobolewski, A.; et al. A Specific Mutation in Muc2 Determines Early Dysbiosis in Colitis-Prone Winnie Mice. Inflamm. Bowel Dis. 2020, 26, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, I.; Blanc, V.; Davidson, N.O. Characterization of Colorectal Cancer Development in Apc (min/+) Mice. Methods Mol. Biol. 2016, 1422, 309–327. [Google Scholar] [PubMed]
- Shoemaker, A.R.; Gould, K.A.; Luongo, C.; Moser, A.R.; Dove, W.F. Studies of neoplasia in the Min mouse. Biochim. Biophys. Acta 1997, 1332, F25–F48. [Google Scholar]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef]
- Ottman, N.; Huuskonen, L.; Reunanen, J.; Boeren, S.; Klievink, J.; Smidt, H.; Belzer, C.; de Vos, W.M. Characterization of Outer Membrane Proteome of Akkermansia muciniphila Reveals Sets of Novel Proteins Exposed to the Human Intestine. Front. Microbiol. 2016, 7, 1157. [Google Scholar] [CrossRef]
- Kang, C.S.; Ban, M.; Choi, E.J.; Moon, H.G.; Jeon, J.S.; Kim, D.K.; Park, S.K.; Jeon, S.G.; Roh, T.Y.; Myung, S.J.; et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Mori, G.; Rampelli, S.; Orena, B.S.; Rengucci, C.; De Maio, G.; Barbieri, G.; Passardi, A.; Casadei Gardini, A.; Frassineti, G.L.; Gaiarsa, S.; et al. Shifts of Faecal Microbiota During Sporadic Colorectal Carcinogenesis. Sci. Rep. 2018, 8, 10329. [Google Scholar] [CrossRef]
- Sobhani, I.; Bergsten, E.; Couffin, S.; Amiot, A.; Nebbad, B.; Barau, C.; de’Angelis, N.; Rabot, S.; Canoui-Poitrine, F.; Mestivier, D.; et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc. Natl. Acad. Sci. USA 2019, 116, 24285–24295. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Hawkins, N.J.; Ward, R.L. Colorectal cancer: A model for epigenetic tumorigenesis. Gut 2007, 56, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Shuwen, H.; Miao, D.; Quan, Q.; Wei, W.; Zhongshan, Z.; Chun, Z.; Xi, Y. Protective effect of the “food-microorganism-SCFAs” axis on colorectal cancer: From basic research to practical application. J. Cancer Res. Clin. Oncol. 2019, 145, 2169–2197. [Google Scholar] [CrossRef]
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Corrigendum: Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional Keys for Intestinal Barrier Modulation. Front. Immunol. 2015, 6, 612. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- McEneany, V.L.; Coyne, M.J.; Chatzidaki-Livanis, M.; Comstock, L.E. Acquisition of MACPF domain-encoding genes is the main contributor to LPS glycan diversity in gut Bacteroides species. ISME J. 2018, 12, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Angrisano, T.; Pero, R.; Peluso, S.; Keller, S.; Sacchetti, S.; Bruni, C.B.; Chiariotti, L.; Lembo, F. LPS-induced IL-8 activation in human intestinal epithelial cells is accompanied by specific histone H3 acetylation and methylation changes. BMC Microbiol. 2010, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, R.W.; Huang, H.; Fletcher, A.; Yu, L.; Pham, Q.; Yu, L.; He, Q.; Wang, T.T.Y. Inhibition of Tumor Growth by Dietary Indole-3-Carbinol in a Prostate Cancer Xenograft Model May Be Associated with Disrupted Gut Microbial Interactions. Nutrients 2019, 11, 467. [Google Scholar] [CrossRef]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Liso, M.; De Santis, S.; Scarano, A.; Verna, G.; Dicarlo, M.; Galleggiante, V.; Campiglia, P.; Mastronardi, M.; Lippolis, A.; Vacca, M.; et al. A Bronze-Tomato Enriched Diet Affects the Intestinal Microbiome under Homeostatic and Inflammatory Conditions. Nutrients 2018, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Ferrocino, I.; Calabrese, F.M.; De Filippis, F.; Cavallo, N.; Siragusa, S.; Rampelli, S.; Di Cagno, R.; Rantsiou, K.; Vannini, L.; et al. Diet influences the functions of the human intestinal microbiome. Sci. Rep. 2020, 10, 4247. [Google Scholar] [CrossRef] [PubMed]
- Scarano, A.; Butelli, E.; De Santis, S.; Cavalcanti, E.; Hill, L.; De Angelis, M.; Giovinazzo, G.; Chieppa, M.; Martin, C.; Santino, A. Combined Dietary Anthocyanins, Flavonols, and Stilbenoids Alleviate Inflammatory Bowel Disease Symptoms in Mice. Front. Nutr. 2017, 4, 75. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Gondalia, S.V.; Palombo, E.A.; Knowles, S.R.; Cox, S.B.; Meyer, D.; Austin, D.W. Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res. 2012, 5, 419–427. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice Genotype | Taxon | coef | pval | qval | IQR Wi_APCMin | IQR Wi | IQR APCMin | IQR WT | |
|---|---|---|---|---|---|---|---|---|---|
| Phylum | Wi_APCMin | Deferribacteres | − | 0.0003 | 0.0031 | 0.06–0.13 | 0.01–0.13 | 1.94–4.34 | 2.67–3.81 |
| Proteobacteria | − | 0.0022 | 0.0123 | 5.3–7.34 | 2.58–6.69 | 16.73–19.03 | 9.84–13.72 | ||
| Verrucomicrobia | + | 0.0004 | 0.0035 | 3.06–6.9 | 0.46–2.94 | 0.11–0.23 | 0.07–0.42 | ||
| Winnie | Deferribacteres | - | 0.0002 | 0.0031 | 0.06–0.13 | 0.01–0.13 | 1.94–4.34 | 2.67–3.81 | |
| Proteobacteria | − | 0.0006 | 0.0037 | 5.3–7.34 | 2.58–6.69 | 16.73–19.03 | 9.84–13.72 | ||
| Family | Wi_APCMin | Acholeplasmataceae | + | 0.0002 | 0.0034 | 0.37–1.12 | 0.36–0.57 | 0–0.01 | 0.01–0.04 |
| Bacteroidaceae | + | 0.0045 | 0.0187 | 8.13–15.14 | 3.35–5.61 | 1.26–4.49 | 1.01–3.56 | ||
| Clostridiaceae_1 | − | 0.0006 | 0.0045 | 0.11–0.22 | 0.35–0.58 | 0.51–1.83 | 0.24–0.46 | ||
| Clostridiaceae_2 | + | 0.0027 | 0.0134 | 0.93–2.08 | 1.63–3.35 | 0.18–0.24 | 0.27–0.89 | ||
| Coriobacteriaceae | − | 0.0028 | 0.0134 | 0.06–0.12 | 0.37–0.73 | 0.25–0.45 | 0.12–0.16 | ||
| Deferribacteraceae | − | 0.0003 | 0.0034 | 0.06–0.13 | 0.01–0.13 | 2.08–4.57 | 2.83–3.98 | ||
| Eubacteriaceae | + | <0.0001 | 0.0031 | 0.61–1.16 | 2.48–4.23 | 0.03–0.07 | 0.08–0.09 | ||
| Helicobacteraceae | − | 0.0053 | 0.0205 | 0.92–2.89 | 0.43–2.47 | 9.17–15.1 | 5.31–9.97 | ||
| Lactobacillaceae | − | 0.0002 | 0.0034 | 0.28–0.56 | 0.87–1.47 | 1.46–4.42 | 1.46–2.04 | ||
| Prevotellaceae | + | 0.0017 | 0.0100 | 12.95–20.68 | 9.23–12.92 | 1.02–6.65 | 1.99–4.92 | ||
| Verrucomicrobiaceae | + | 0.0008 | 0.0054 | 3.17–7.15 | 0.49–3.11 | 0.07–0.21 | 0.01–0.36 | ||
| Winnie | Acholeplasmataceae | + | 0.0005 | 0.0042 | 0.37–1.12 | 0.36–0.57 | 0–0.01 | 0.01–0.04 | |
| Clostridiaceae_2 | + | 0.0005 | 0.0043 | 0.93–2.08 | 1.63–3.35 | 0.18–0.24 | 0.27–0.89 | ||
| Deferribacteraceae | − | 0.0003 | 0.0034 | 0.06–0.13 | 0.01–0.13 | 2.08–4.57 | 2.83–3.98 | ||
| Enterobacteriaceae | − | 0.0039 | 0.0170 | 0.02–0.17 | 0.01–0.01 | 0.09–0.49 | 0.01–0.04 | ||
| Eubacteriaceae | + | <0.0001 | 0.0003 | 0.61–1.16 | 2.48–4.23 | 0.03–0.07 | 0.08–0.09 | ||
| Helicobacteraceae | − | 0.0018 | 0.0100 | 0.92–2.89 | 0.43–2.47 | 9.17–15.1 | 5.31–9.97 | ||
| Genus | Wi_APCMin | Acholeplasma | + | 0.0002 | 0.0037 | 0.39–1.19 | 0.38–0.58 | 0–0.01 | 0.01–0.04 |
| Akkermansia | + | 0.0007 | 0.0066 | 3.36–7.52 | 0.52–3.29 | 0.06–0.22 | 0.01–0.38 | ||
| Alkaliphilus | + | 0.0016 | 0.0124 | 0.97–2.18 | 1.71–3.33 | 0.1–0.23 | 0.25–0.89 | ||
| Alloprevotella | + | <0.0001 | <0.0001 | 1.59–2.55 | 0.98–2.52 | 0.01–0.1 | 0–0.01 | ||
| Bacteroides | + | 0.0045 | 0.0267 | 8.63–15.00 | 3.52–6.01 | 1.33–4.66 | 1.07–3.79 | ||
| Clostridium sensu stricto | − | 0.0007 | 0.0066 | 0.11–0.21 | 0.34–0.54 | 0.51–1.85 | 0.23–0.48 | ||
| Eubacterium | + | <0.0001 | 0.0008 | 0.62–1.18 | 2.6–4.28 | 0.03–0.05 | 0.05–0.07 | ||
| Helicobacter | − | 0.0059 | 0.0323 | 0.98–3.05 | 0.41–2.62 | 9.41–15.65 | 5.67–10.43 | ||
| Lactobacillus | − | 0.0002 | 0.0037 | 0.3–0.59 | 0.92–1.51 | 1.47–4.62 | 1.55–2.12 | ||
| Mucispirillum | − | 0.0003 | 0.0041 | 0.06–0.14 | 0.01–0.14 | 2.11–4.71 | 2.92–4.24 | ||
| Odoribacter | − | 0.0023 | 0.0155 | 0.99–1.42 | 0.71–1.6 | 2.22–4.5 | 3.26–3.65 | ||
| Paraprevotella | + | <0.0001 | 0.0012 | 0.80–2.87 | 1.07–2.15 | 0.01–0.06 | 0.02–0.09 | ||
| Porphyromonas | + | 0.0021 | 0.0150 | 0.41–0.64 | 0.39–0.92 | 0.09–0.2 | 0.05–0.09 | ||
| Prevotella | + | 0.0038 | 0.0242 | 9.11–15.07 | 6.07–8.24 | 0.62–6.54 | 1.96–5 | ||
| Rikenella | − | 0.0002 | 0.0037 | 0.06–0.45 | 0.49–1.01 | 2.92–5.48 | 1.53–3.8 | ||
| Stomatobaculum | − | 0.0009 | 0.0077 | 0.08–0.15 | 0.18–0.57 | 1.28–3.33 | 2.18–5.3 | ||
| Syntrophococcus | − | 0.0080 | 0.0418 | 0.01–0.06 | 0.02–0.05 | 0.2–0.59 | 0.13–0.69 | ||
| Ureaplasma | − | 0.0006 | 0.0066 | 0–0.04 | 0.01–0.06 | 0.28–0.57 | 0.14–0.25 | ||
| Winnie | Acholeplasma | + | 0.0005 | 0.0053 | 0.39–1.19 | 0.38–0.58 | 0–0.01 | 0.01–0.04 | |
| Alkaliphilus | + | 0.0003 | 0.0038 | 0.97–2.18 | 1.71–3.33 | 0.1–0.23 | 0.25–0.89 | ||
| Alloprevotella | + | <0.0001 | 0.0003 | 1.59–2.55 | 0.98–2.52 | 0.01–0.1 | 0–0.01 | ||
| Escherichia/Shigella | − | 0.0082 | 0.0418 | 0.01–0.17 | n.d.* | 0.09–0.49 | 0.01–0.04 | ||
| Eubacterium | + | <0.0001 | 0.0001 | 0.62–1.18 | 2.6–4.28 | 0.03–0.05 | 0.05–0.07 | ||
| Helicobacter | − | 0.0019 | 0.0137 | 0.98–3.05 | 0.41–2.62 | 9.41–15.65 | 5.67–10.43 | ||
| Mucispirillum | − | 0.0003 | 0.0038 | 0.06–0.14 | 0.01–0.14 | 2.11–4.71 | 2.92–4.24 | ||
| Odoribacter | − | 0.0052 | 0.0293 | 0.99–1.42 | 0.71–1.6 | 2.22–4.5 | 3.26–3.65 | ||
| Paraprevotella | + | 0.0002 | 0.0037 | 0.80–2.87 | 1.07–2.15 | 0.01–0.06 | 0.02–0.09 | ||
| Porphyromonas | + | 0.0014 | 0.0115 | 0.41–0.64 | 0.39–0.92 | 0.09–0.2 | 0.05–0.09 | ||
| Ureaplasma | − | 0.0040 | 0.0242 | 0–0.04 | 0.01–0.06 | 0.28–0.57 | 0.14–0.25 |
| OTU | Group | coef | pval | qval |
|---|---|---|---|---|
| Akkermansia muciniphila | Winnie-APCMin/+ | + | 0.0007 | 0.0303 |
| Clostridium leptum (Clostridium cluster IV) | Winnie-APCMin/+ | − | 0.0015 | 0.0455 |
| Eubacterium coprostanoligenes | Winnie-APCMin/+ | + | <0.0001 | 0.0007 |
| Lactobacillus intestinalis | Winnie-APCMin/+ | − | <0.0001 | 0.0079 |
| Mucispirillum schaedleri | Winnie-APCMin/+ | − | 0.0003 | 0.0142 |
| Paraprevotella clara | Winnie-APCMin/+ | + | <0.0001 | 0.0043 |
| Prevotella oralis | Winnie-APCMin/+ | + | <0.0001 | 0.0043 |
| Prevotellamassilia timonensis | Winnie-APCMin/+ | + | <0.0001 | 0.0006 |
| Rikenella microfusus | Winnie-APCMin/+ | − | 0.0002 | 0.0116 |
| Eubacterium coprostanoligenes | Winnie | + | <0.0001 | 0.0004 |
| Lactobacillus intestinalis | Winnie | − | 0.0002 | 0.0120 |
| Mucispirillum schaedleri | Winnie | − | 0.0002 | 0.0122 |
| Paraprevotella clara | Winnie | + | 0.0003 | 0.0142 |
| Prevotellamassilia timonensis | Winnie | + | <0.0001 | 0.0033 |
| Ruminococcus champanellensis | Winnie | + | 0.0010 | 0.0336 |
| Genotype-Tissue (n. Mice) | Non Dysplastic ACF | Dysplastic ACF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Groups | Unicryptic Lesion | Microadenoma >1 ≤5—LG | Microadenoma >5—LG | Microadenoma >5—HG | ||||||||
| Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | |
| APCMin/+—PC (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+—MC (2) | 50 | 0.5 ± 0.5 | 100 | 1.5 ± 0.5 | 50 | 0.5 ± 0.5 | 0 | 0 | 100 | 1.0 ± 0 | 0 | 0 |
| APCMin/+—DC (2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Genotype Tissue Treatment (n. Mice) | Non Dysplastic ACF | Dysplastic ACF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Groups | Unicryptic Lesion | Microadenoma >1 ≤5 —LG | Microadenoma >5—LG | Microadenoma >5—HG | ||||||||
| Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | Incidence (%) | Multiplicity (Mean ± SEM) | |
| APCMin/+—PC with FMT from WT (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+—PC with FMT from Winnie (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+—MC with FMT from WT (3) | 33.3 | 0.33 ± 0.33 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+—MC with FMT from Winnie (7) | 14.3 | 0.14 ± 0.14 | 42.6 | 0.43 ± 0.2 | 0 | 0 | 28.6 | 0.29 ± 0.19 | 14.3 | 0.14 ± 0.14 | 0 | 0 |
| APCMin/+—DC with FMT from WT (3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+—DC with FMT from Winnie (7) | 14.3 | 0.86 ± 0.86 | 42.6 | 0.86 ± 0.6 | 14.3 | 0.14 ± 0.14 | 14.3 | 0.14 ± 0.14 | 28.6 | 0.14 ± 0.37 | 28.6 | 0.29 ± 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Santis, S.; Liso, M.; Vacca, M.; Verna, G.; Cavalcanti, E.; Coletta, S.; Calabrese, F.M.; Eri, R.; Lippolis, A.; Armentano, R.; et al. Dysbiosis Triggers ACF Development in Genetically Predisposed Subjects. Cancers 2021, 13, 283. https://doi.org/10.3390/cancers13020283
De Santis S, Liso M, Vacca M, Verna G, Cavalcanti E, Coletta S, Calabrese FM, Eri R, Lippolis A, Armentano R, et al. Dysbiosis Triggers ACF Development in Genetically Predisposed Subjects. Cancers. 2021; 13(2):283. https://doi.org/10.3390/cancers13020283
Chicago/Turabian StyleDe Santis, Stefania, Marina Liso, Mirco Vacca, Giulio Verna, Elisabetta Cavalcanti, Sergio Coletta, Francesco Maria Calabrese, Rajaraman Eri, Antonio Lippolis, Raffaele Armentano, and et al. 2021. "Dysbiosis Triggers ACF Development in Genetically Predisposed Subjects" Cancers 13, no. 2: 283. https://doi.org/10.3390/cancers13020283
APA StyleDe Santis, S., Liso, M., Vacca, M., Verna, G., Cavalcanti, E., Coletta, S., Calabrese, F. M., Eri, R., Lippolis, A., Armentano, R., Mastronardi, M., De Angelis, M., & Chieppa, M. (2021). Dysbiosis Triggers ACF Development in Genetically Predisposed Subjects. Cancers, 13(2), 283. https://doi.org/10.3390/cancers13020283