Multi-Omics and Informatics Analysis of FFPE Tissues Derived from Melanoma Patients with Long/Short Responses to Anti-PD1 Therapy Reveals Pathways of Response

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patient and Tumor Characteristics
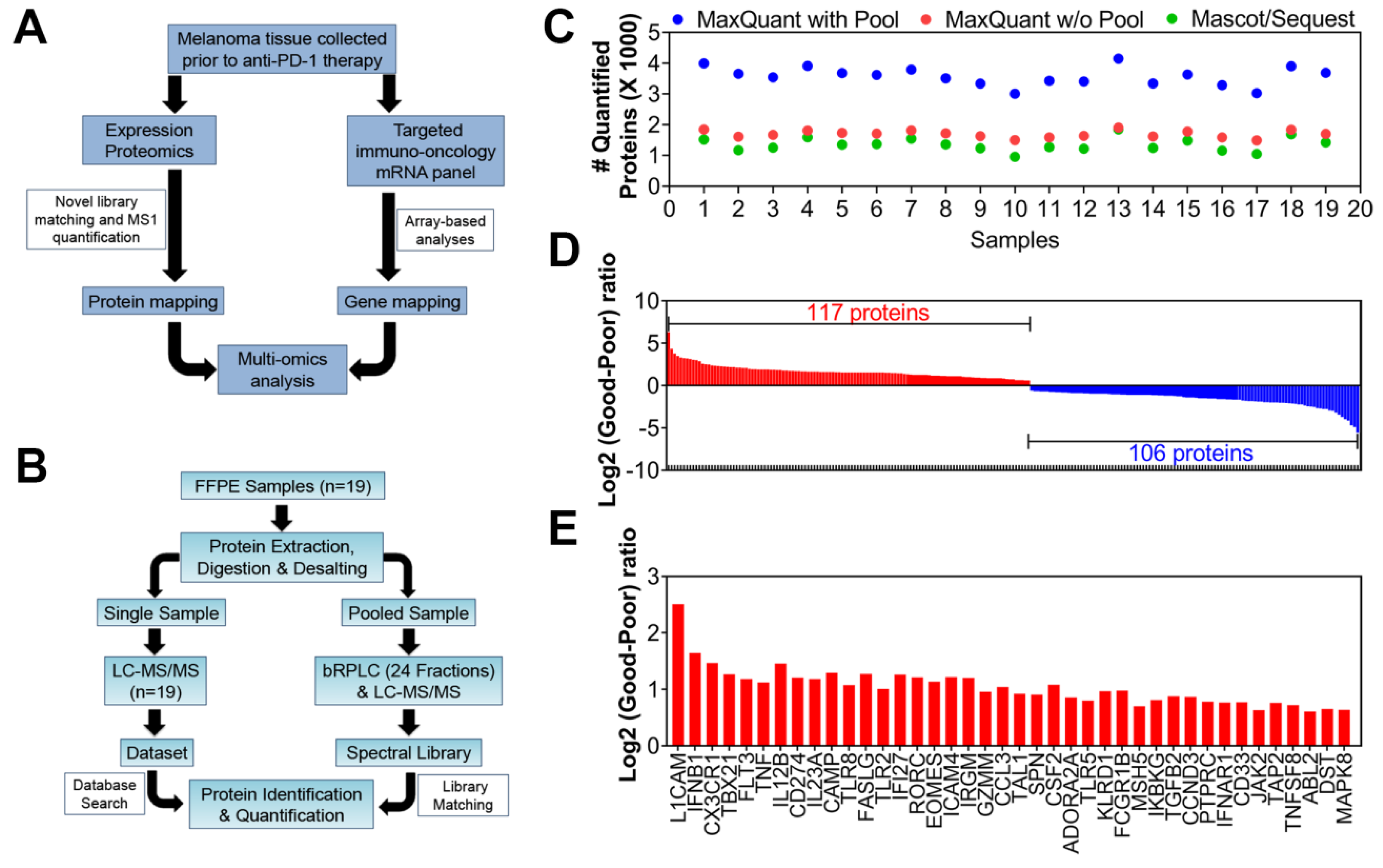
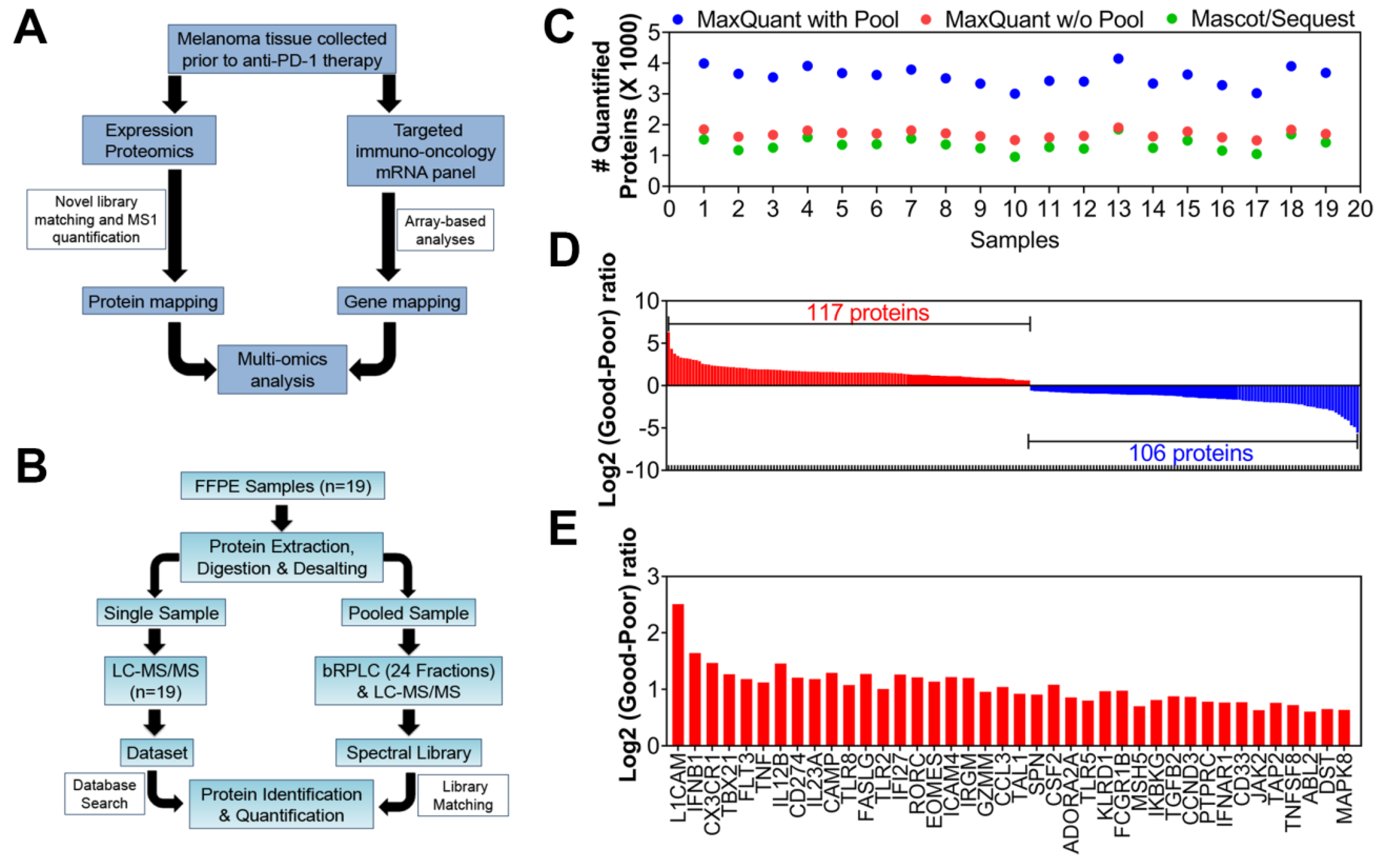
2.2. Multi-Omics Studies: Proteomics and Targeted-mRNA Analyses
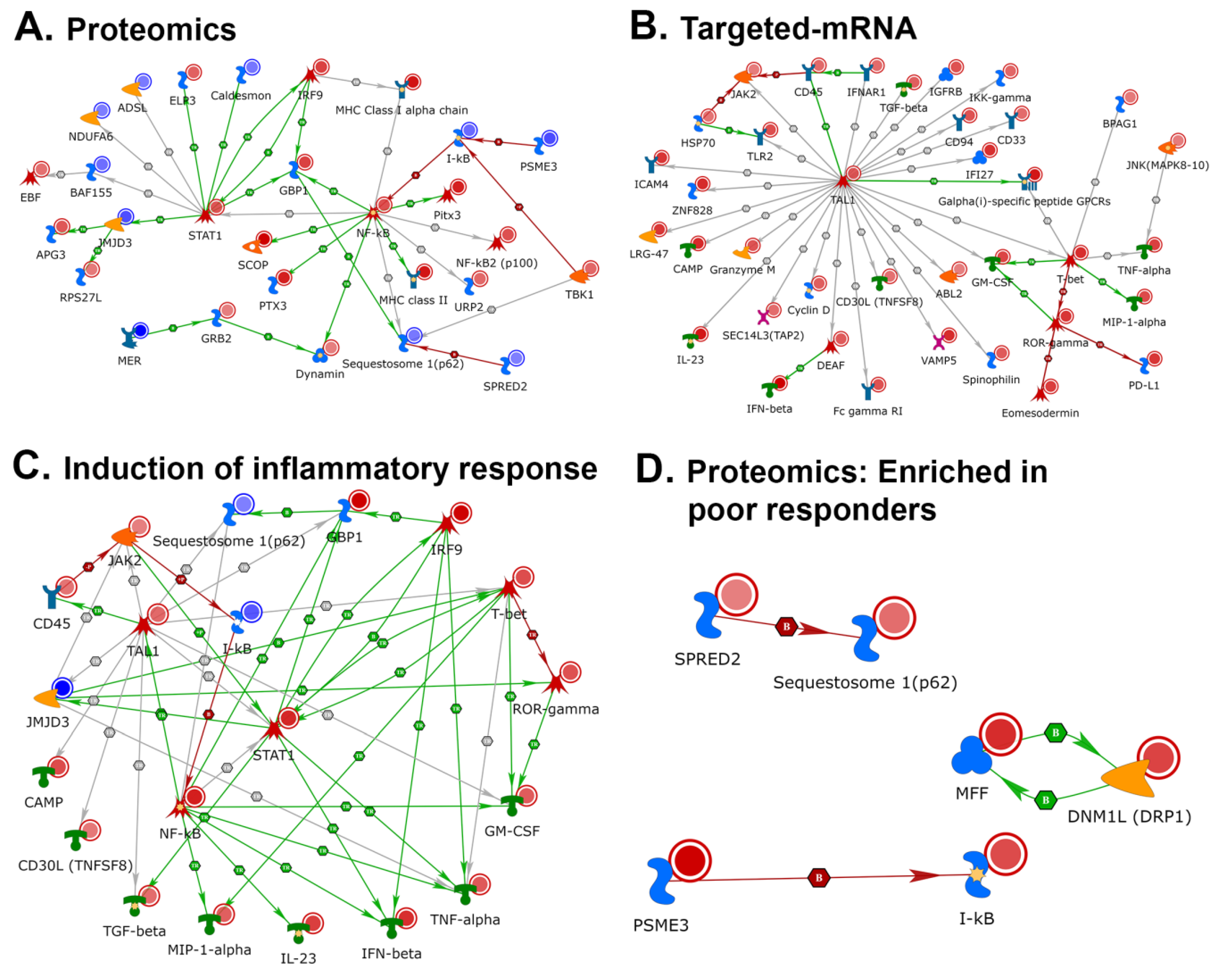
2.3. Network Analyses of Differentially Expressed Candidates
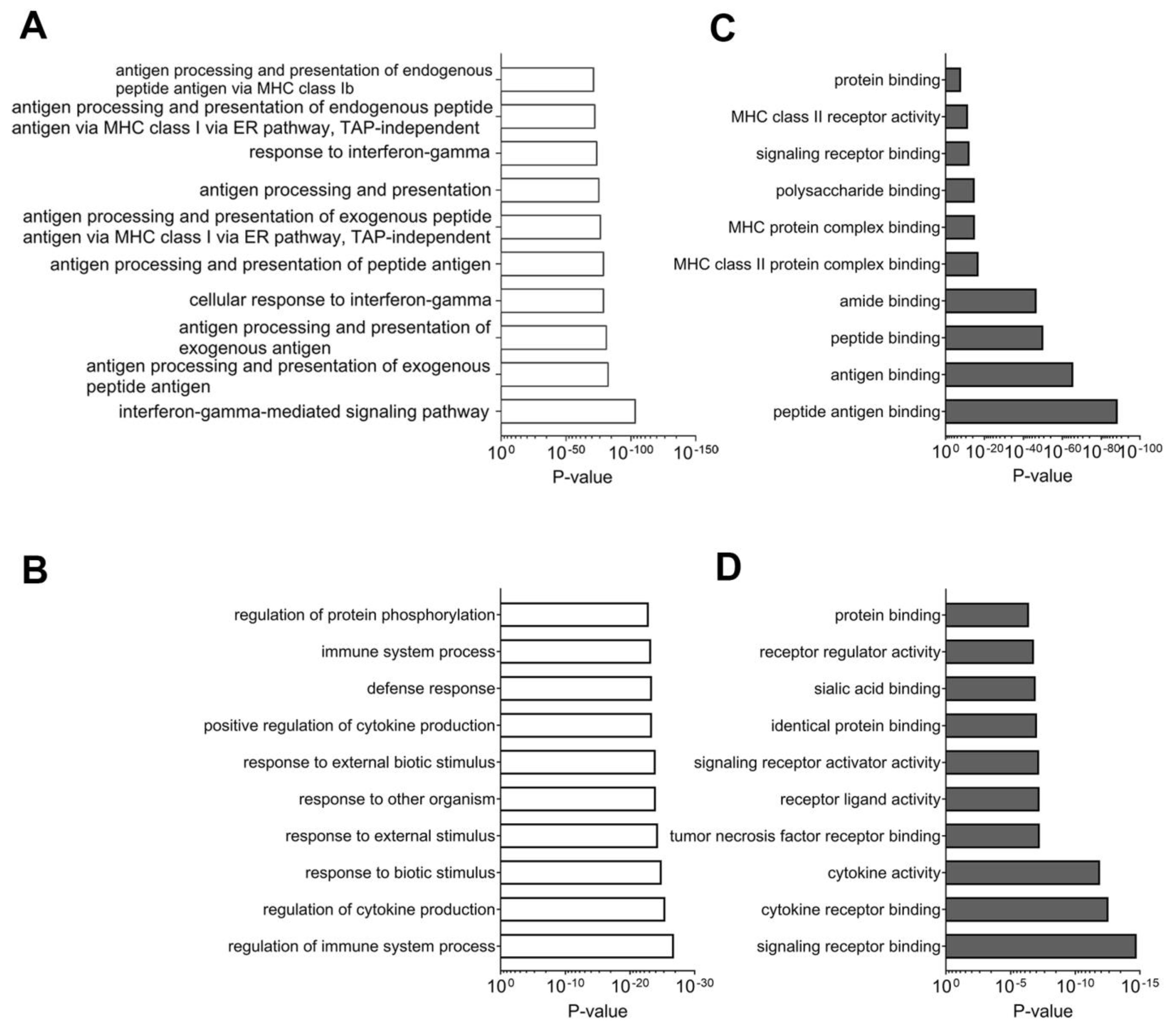
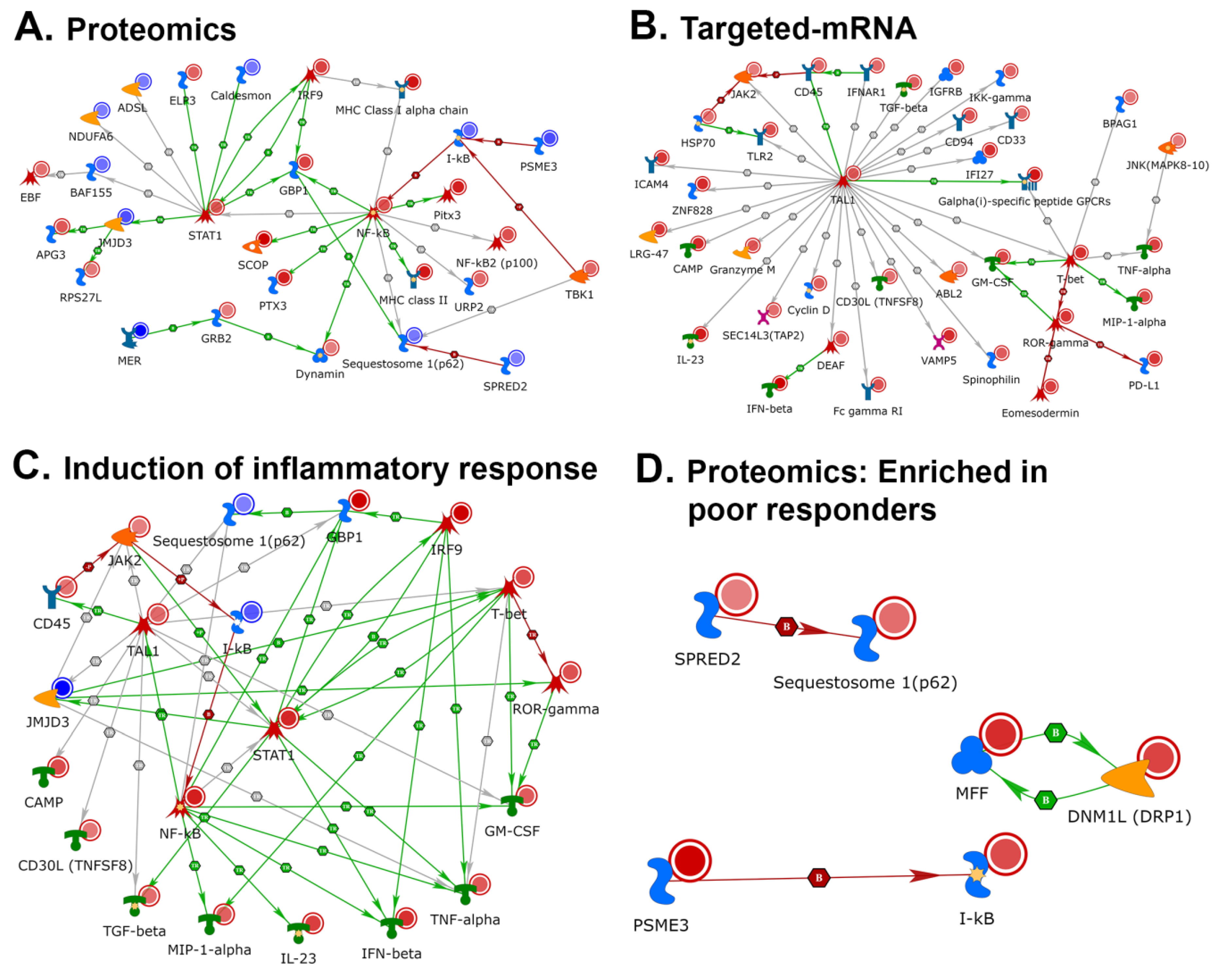
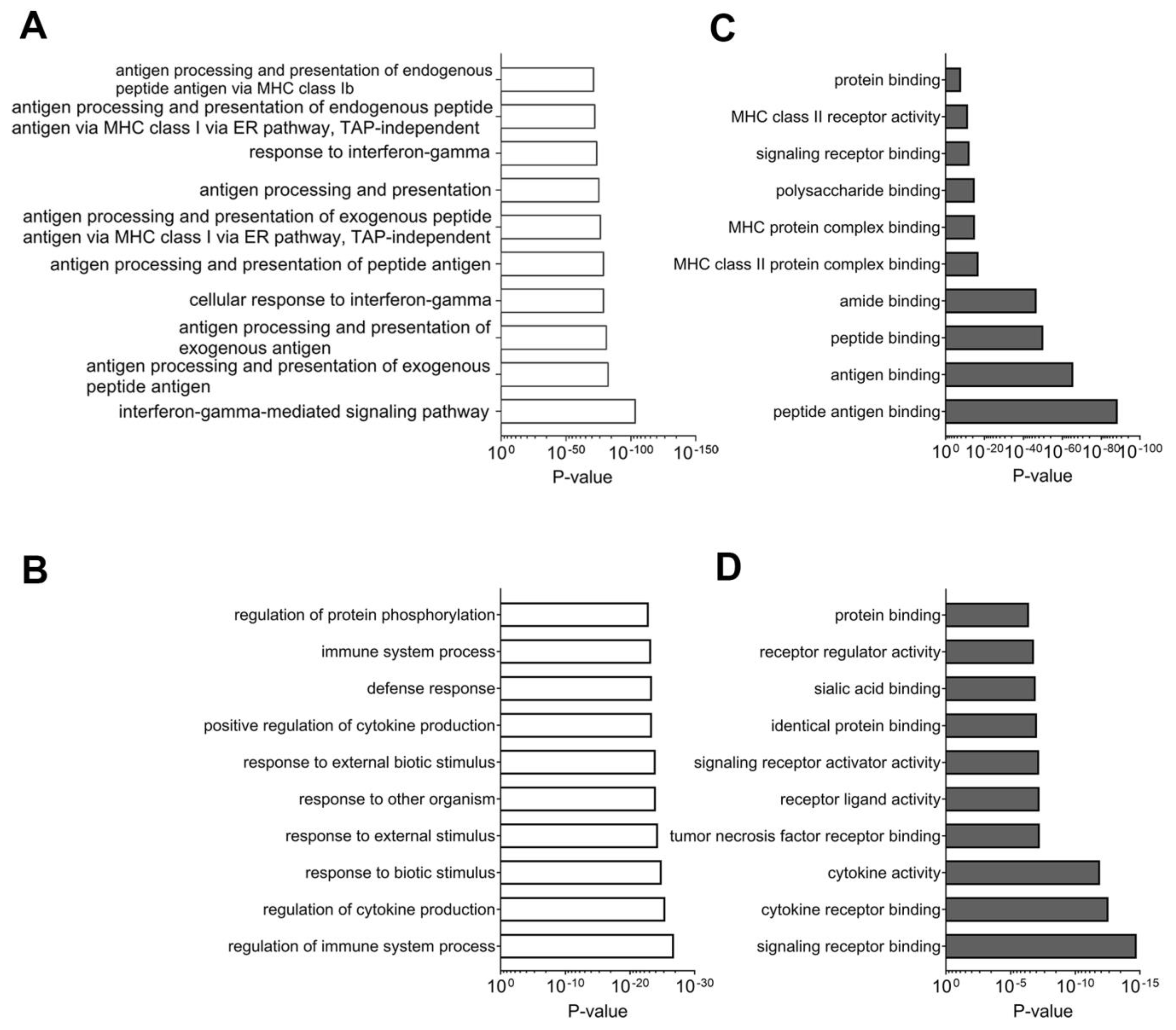
2.3.1. Network Analyses of Differentially Expressed Genes and Proteins: Multi-Omics Datasets Analyzed Individually for Processes and Pathways Enriched in Tissues Samples from Good Responders to Anti-PD-1 Therapy
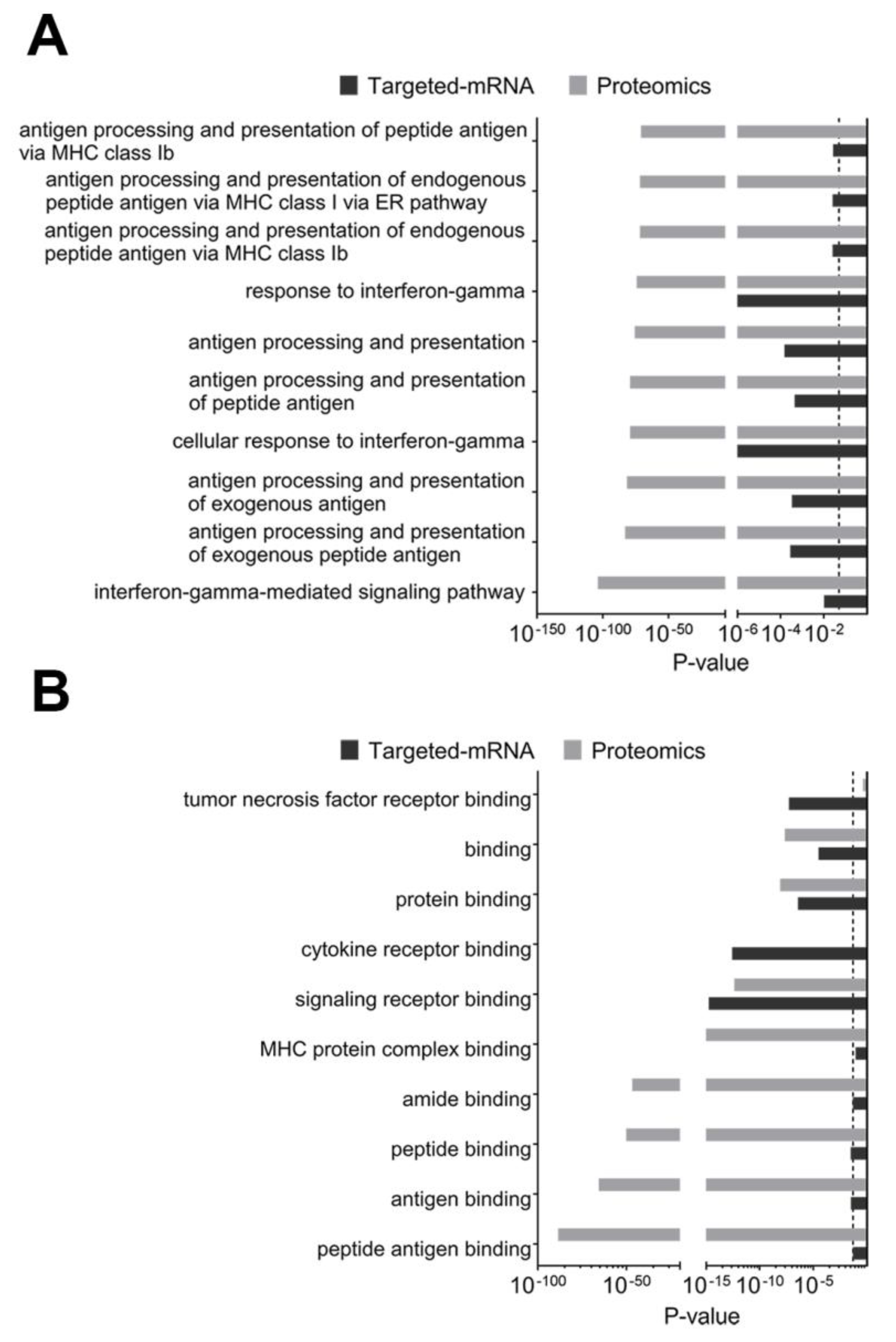
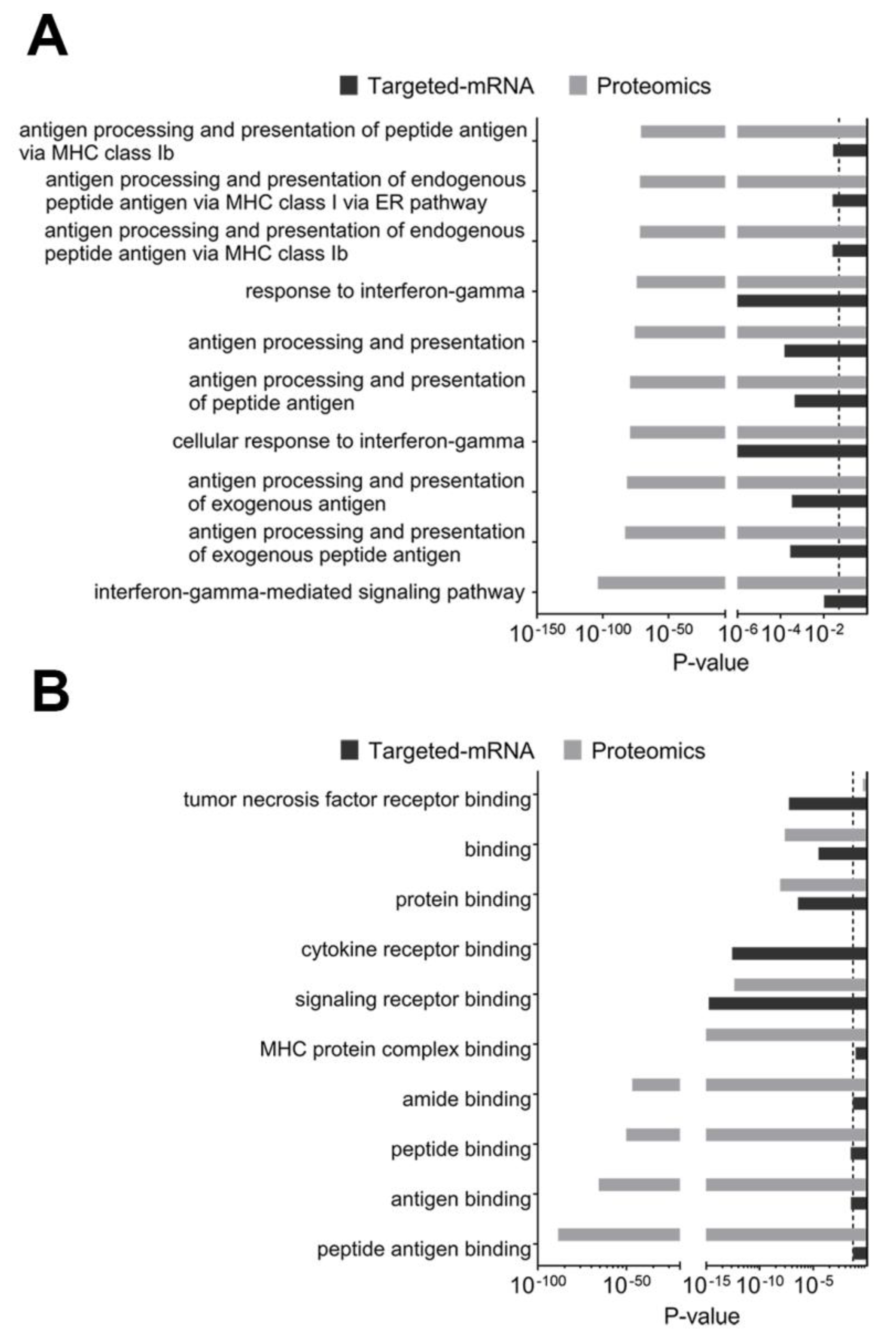
2.3.2. Composite Analyses of Multi-Omics Data (Proteomics Combined with Targeted Transcriptomics) Showed the Importance of Antigen Presentation
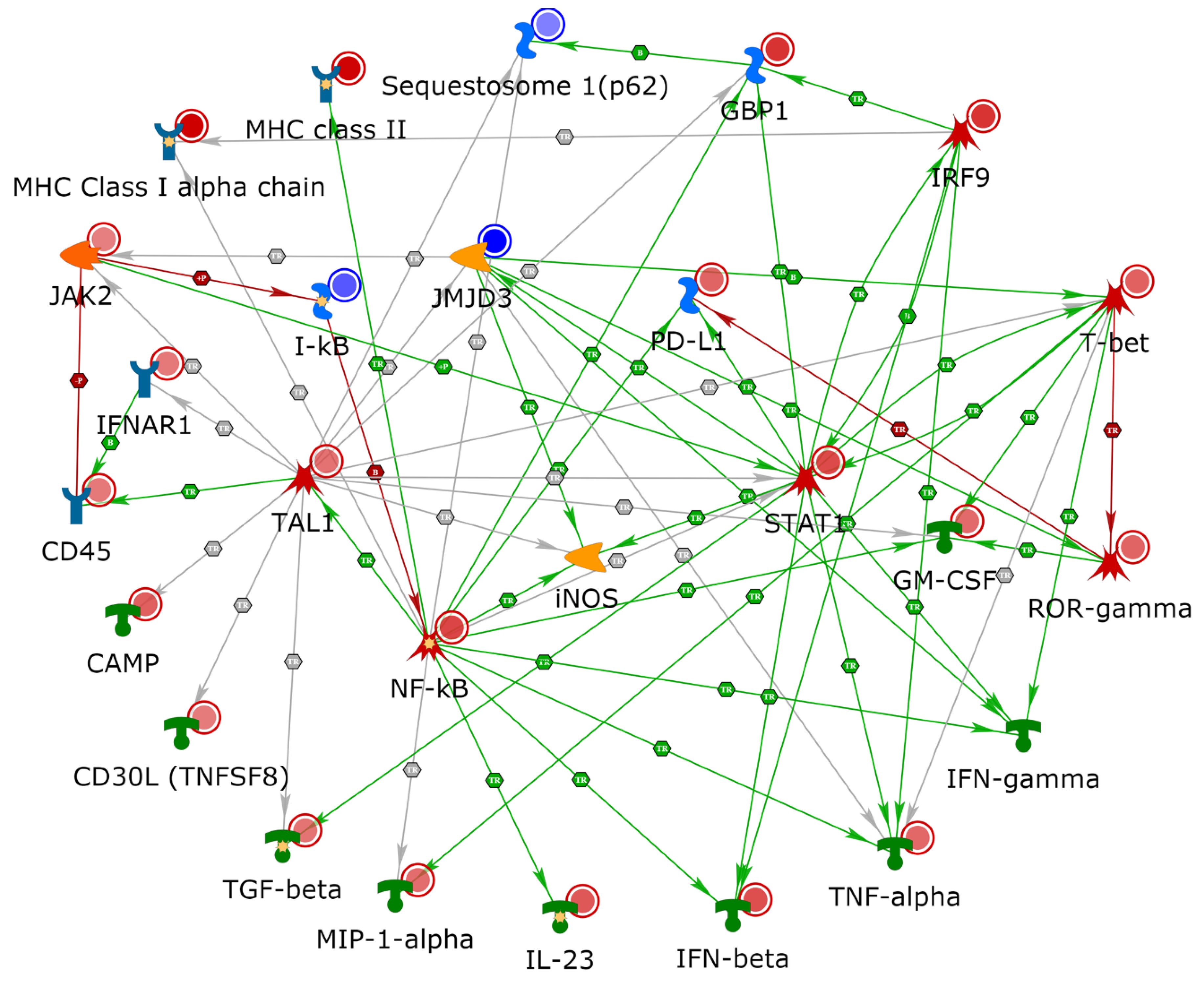
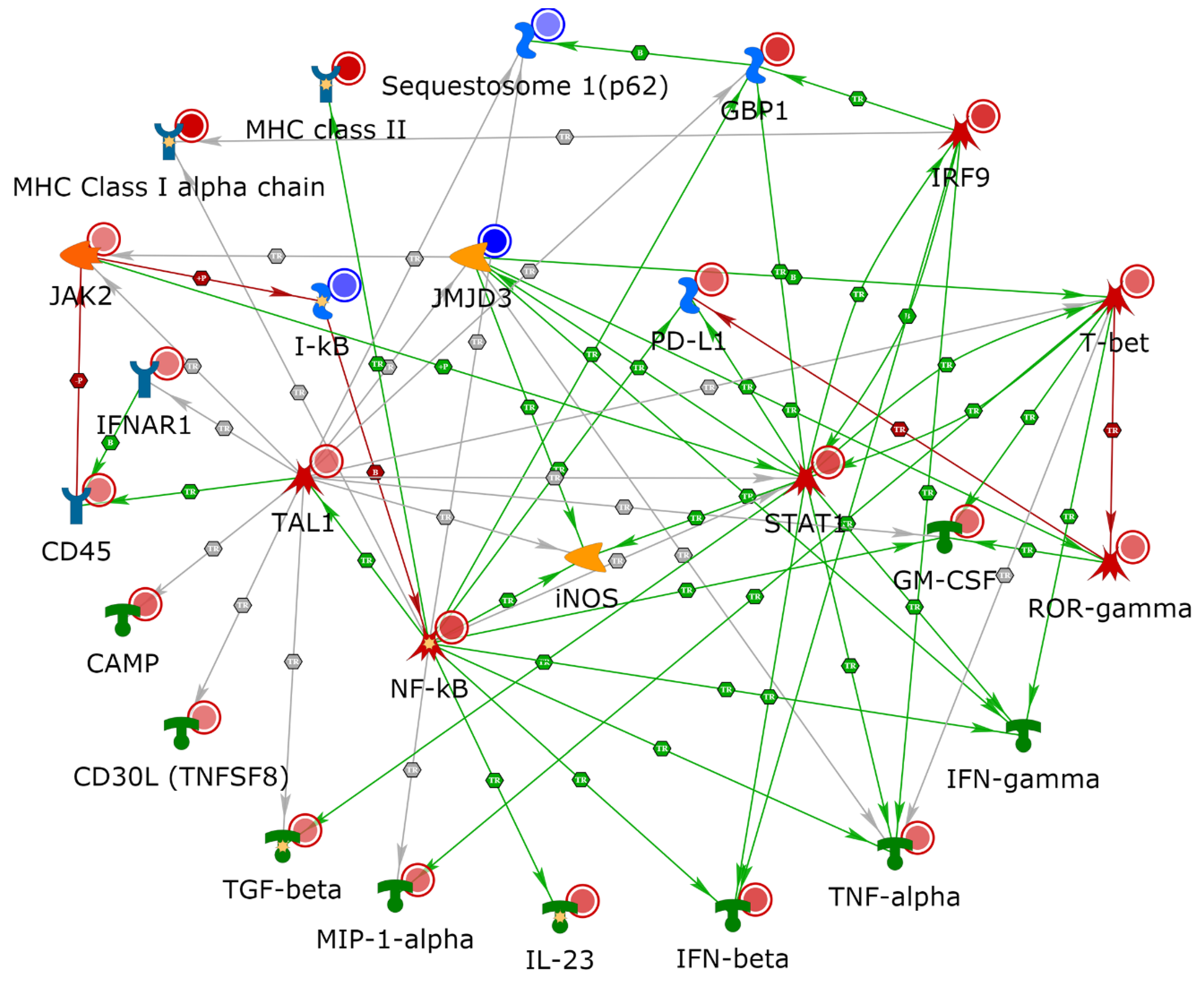
2.4. NETWORK Analyses Illustrates Players of Antigen Presentation Pathways
2.5. Use of TIMER2.0 to Determine Immune Cell Populations in Patient Specimens
3. Discussion
4. Materials and Methods
4.1. Collection of FFPE Samples and Clinical Information
4.2. Mass Spectrometry Analysis
4.3. Targeted-mRNA Analyses
4.4. TIMER2.0 Analysis
4.5. Analysis Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Zavala, V.A.; Kalergis, A.M. New clinical advances in immunotherapy for the treatment of solid tumours. Immunology 2015, 145, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Moreno, B.H.; Parisi, G.; Robert, L.; Ribas, A. Anti–PD-1 Therapy in Melanoma. Semin. Oncol. 2015, 42, 466–473. [Google Scholar] [CrossRef]
- Jarboe, J.; Gupta, A.; Saif, W. Therapeutic Human Monoclonal Antibodies against Cancer. Methods Mol. Biol. 2014, 1060, 61–77. [Google Scholar] [CrossRef]
- Pennock, G.K.; Chow, L.Q. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Lao, C.D.; Urba, W.J.; McDermott, D.F.; Horak, C.; Jiang, J.; Wolchok, J.D. Efficacy and Safety of Nivolumab in Patients with BRAF V600 Mutant and BRAF Wild-Type Advanced Melanoma: A Pooled Analysis of 4 Clinical Trials. JAMA Oncol. 2015, 1, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Callahan, M.K.; Postow, M.A.; Wolchok, J.D. On being less tolerant: Enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med. 2015, 7, 280sr1. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Fuertes, M.B.; Woo, S.-R. Innate immune sensing of cancer: Clues from an identified role for type I IFNs. Cancer Immunol. Immunother. 2012, 61, 1343–1347. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef]
- Tan, A.C.; Emmett, L.; Lo, S.; Liu, V.; Kapoor, R.; Carlino, M.S.; Guminski, A.D.; Long, G.V.; Menzies, A.M. FDG-PET response and outcome from anti-PD-1 therapy in metastatic melanoma. Ann. Oncol. 2018, 29, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Welsh, E.A.; Eschrich, S.A.; Berglund, A.E.; Fenstermacher, D.A. Iterative rank-order normalization of gene expression microarray data. BMC Bioinform. 2013, 14, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesca, I.; Tunger, A.; Müller, L.; Wehner, R.; Lai, X.; Grimm, M.-O.; Rutella, S.; Bachmann, M.; Schmitz, M. Characteristics of Tumor-Infiltrating Lymphocytes Prior to and During Immune Checkpoint Inhibitor Therapy. Front. Immunol. 2020, 11, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Georganaki, M.; Van Hooren, L.; Dimberg, A. Vascular Targeting to Increase the Efficiency of Immune Checkpoint Blockade in Cancer. Front. Immunol. 2018, 9, 3081. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Longo, V. Angiogenesis and Melanoma. Cancers 2010, 2, 114. [Google Scholar] [CrossRef]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 2020, 4. [Google Scholar] [CrossRef] [Green Version]
- Martin-Broto, J.; Cruz, J.; Penel, N.; Le Cesne, A.; Hindi, N.; Luna, P.; Moura, D.S.; Bernabeu, D.; De Alava, E.; Lopez-Guerrero, J.A.; et al. Pazopanib for treatment of typical solitary fibrous tumours: A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 456–466. [Google Scholar] [CrossRef]
- Aguado, B.A.; Wu, J.J.; Azarin, S.M.; Nanavati, D.; Rao, S.S.; Bushnell, G.G.; Medicherla, C.B.; Shea, L.D. Secretome identification of immune cell factors mediating metastatic cell homing. Sci. Rep. 2015, 5, 17566. [Google Scholar] [CrossRef]
- Kumar, S.U.; Kumar, D.T.; Siva, R.; Doss, C.G.P.; Zayed, H. Integrative Bioinformatics Approaches to Map Potential Novel Genes and Pathways Involved in Ovarian Cancer. Front. Bioeng. Biotechnol. 2019, 7, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Sugimoto, M.; Kawaguchi, K.; Pu, F.; Uozumi, R.; Yamaguchi, A.; Nishie, M.; Tsuda, M.; Kotake, T.; Morita, S.; et al. Gene expression profile of peripheral blood mononuclear cells may contribute to the identification and immunological classification of breast cancer patients. Breast Cancer 2019, 26, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Matikainen, S.; Paananen, A.; Miettinen, M.; Kurimoto, M.; Timonen, T.; Julkunen, I.; Sareneva, T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: Differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur. J. Immunol. 2001, 31, 2236–2245. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Tan, T.H.; Rice, N.; Kretzschmar, M.; Ghosh, P.; Young, H.A. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc. Natl. Acad. Sci. USA 1992, 89, 1740–1744. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56bright subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yang, W.; Pan, M.; Scully, E.; Girardi, M.; Augenlicht, L.H.; Craft, J.; Yin, Z. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 2003, 198, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Leite-De-Moraes, M.C.; Moreau, G.; Arnould, A.; Machavoine, F.; Garcia, C.; Papiernik, M.; Dy, M. IL-4-producing NK T cells are biased towards IFN-gamma production by IL-12. Influence of the microenvironment on the functional capacities of NK T cells. Eur. J. Immunol. 1998, 28, 1507–1515. [Google Scholar] [CrossRef]
- Matsushita, H.; Hosoi, A.; Ueha, S.; Abe, J.; Fujieda, N.; Tomura, M.; Maekawa, R.; Matsushima, K.; Ohara, O.; Kakimi, K. Cytotoxic T Lymphocytes Block Tumor Growth Both by Lytic Activity and IFNgamma-Dependent Cell-Cycle Arrest. Cancer Immunol. Res. 2015, 3, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.P.; Goodrich, S.; Gerth, A.J.; Peng, S.L.; Lund, F.E. Regulation of IFN-gamma Production by B Effector 1 Cells: Essential Roles for T-bet and the IFN-gamma Receptor. J. Immunol. 2005, 174, 6781–6790. [Google Scholar] [CrossRef] [Green Version]
- Olalekan, S.A.; Cao, Y.; Hamel, K.M.; Finnegan, A. B cells expressing IFN-gamma suppress Treg-cell differentiation and promote autoimmune experimental arthritis. Eur. J. Immunol. 2015, 45, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Okamura, H.; Tagawa, Y.I.; Iwakura, Y.; Nakanishi, K. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-gamma production from activated B cells. Proc. Natl. Acad. Sci USA 1997, 94, 3948–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. 2016, 76, 5597–5601. [Google Scholar] [CrossRef] [Green Version]
- Messina, J.L.; Fenstermacher, D.A.; Eschrich, S.; Qu, X.; Berglund, A.E.; Lloyd, M.C.; Schell, M.J.; Sondak, V.K.; Weber, J.S.; Mulé, J.J. 12-Chemokine Gene Signature Identifies Lymph Node-like Structures in Melanoma: Potential for Patient Selection for Immunotherapy? Sci. Rep. 2012, 2, 765. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.F.; Horvath, C.M.; Wen, Z.; Schreiber, R.D.; Darnell, J.E., Jr. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 7673–7678. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.E.; Kitagawa, M.; Su, W.C.; You, Z.H.; Iwamoto, Y.; Fu, X.Y. Cell Growth Arrest and Induction of Cyclin-Dependent Kinase Inhibitor p21WAF1/CIP1 Mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef]
- Ellis, T.N.; Beaman, B.L. Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology 2004, 112, 2–12. [Google Scholar] [CrossRef]
- Harvat, B.L.; Seth, P.; Jetten, A.M. The role of p27Kip1 in gamma interferon-mediated growth arrest of mammary epithelial cells and related defects in mammary carcinoma cells. Oncogene 1997, 14, 2111–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Mauldin, I.S.; Wages, N.A.; Stowman, A.M.; Wang, E.; Smolkin, M.E.; Olson, W.C.; Deacon, D.H.; Smith, K.T.; Galeassi, N.V.; Chianese-Bullock, K.A.; et al. Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol. Immunother. 2016, 65, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.; James, E. Antigen processing and immune regulation in the response to tumours. Immunology 2017, 150, 16–24. [Google Scholar] [CrossRef] [Green Version]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-W.; Lee, Y.; Kim, D.-S.; Kwon, H.-J. Direct role of NF-kappaB activation in Toll-like receptor-triggered HLA-DRA expression. Eur. J. Immunol. 2006, 36, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Steimle, V.; Siegrist, C.A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265, 106–109. [Google Scholar] [CrossRef]
- Ozao-Choy, J.; Lee, D.J.; Faries, M.B. Melanoma Vaccines: Mixed past, promising future. Surg. Clin. North Am. 2014, 94, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Maurer, D.M.; Butterfield, L.H.; Vujanovic, L. Melanoma vaccines: Clinical status and immune endpoints. Melanoma Res. 2019, 29, 109–118. [Google Scholar] [CrossRef]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef]
- Terme, J.-M.; Wencker, M.; Favre-Bonvin, A.; Bex, F.; Gazzolo, L.; Dodon, M.D.; Jalinot, P. Cross Talk between Expression of the Human T-Cell Leukemia Virus Type 1 Tax Transactivator and the Oncogenic bHLH Transcription Factor TAL1. J. Virol. 2008, 82, 7913–7922. [Google Scholar] [CrossRef] [Green Version]
- Vagapova, E.R.; Spirin, P.V.; Lebedev, T.D.; Prassolov, V.S. The Role of TAL1 in Hematopoiesis and Leukemogenesis. Acta Nat. 2018, 10, 15–23. [Google Scholar] [CrossRef]
- Hoang, T.; Lambert, J.A.; Martin, R. SCL/TAL1 in Hematopoiesis and Cellular Reprogramming. Curr. Top. Dev. Biol. 2016, 118, 163–204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kurukuti, S.; Saffrey, P.; Vukovic, M.; Michie, A.M.; Strogantsev, R.; West, A.G.; Vetrie, D. Chromatin looping defines expression of TAL1, its flanking genes, and regulation in T-ALL. Blood 2013, 122, 4199–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, W.-S.; Ngoc, P.C.; Sanda, T. Roles of the RUNX1 Enhancer in Normal Hematopoiesis and Leukemogenesis. Adv. Exp. Med. Biol. 2017, 962, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Massagué, J. TGF-beta Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.R.; Bastin, D.; Chen, L.; Nguyen, A.; Storbeck, C.J.; Lefebvre, C.; Stojdl, D.; Bramson, J.L.; Bell, J.C.; Wan, Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Investig. 2019, 129, 518–530. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, Efficacy, and Biomarkers of Nivolumab With Vaccine in Ipilimumab-Refractory or -Naive Melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment. Cell Melanoma Res. 2014, 28, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, T.; Ward, J.P.; Gubin, M.M.; Arthur, C.D.; Lee, S.H.; Hundal, J.; Selby, M.J.; Graziano, R.F.; Mardis, E.R.; Korman, A.J.; et al. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol. Res. 2017, 5, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Markowitz, J. The use of baseline biomarkers to predict outcome in melanoma patients treated with pembrolizumab. Ann. Res. Hosp. 2017, 1. [Google Scholar] [CrossRef]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [Green Version]
- Ozgun, A.; Sondak, V.K.; Markowitz, J. Resistance patterns to anti-PD-1 therapy in metastatic melanoma. Chin. Clin. Oncol. 2016, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Karras, P.; Riveiro-Falkenbach, E.; Cañón, E.; Tejedo, C.; Calvo, T.G.; Martínez-Herranz, R.; Alonso-Curbelo, D.; Cifdaloz, M.; Perez-Guijarro, E.; Gómez-López, G.; et al. p62/SQSTM1 Fuels Melanoma Progression by Opposing mRNA Decay of a Selective Set of Pro-metastatic Factors. Cancer Cell 2019, 35, 46–63.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.H.; Chae, Y.C.; Kossenkov, A.V.; Lee, Y.G.; Tang, H.-Y.; Agarwal, E.; Gabrilovich, D.I.; Languino, L.R.; Speicher, D.W.; Shastrula, P.K.; et al. MFF Regulation of Mitochondrial Cell Death Is a Therapeutic Target in Cancer. Cancer Res. 2019, 79, 6215–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuyoshi, H.; Orisaka, M.; Fujita, Y.; Asare-Werehene, M.; Tsang, B.K.; Yoshida, Y. Prognostic impact of Dynamin related protein 1 (Drp1) in epithelial ovarian cancer. BMC Cancer 2020, 20, 467. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Pan, F.; Li, S.; Huang, R.; Wang, X.; Wang, S.; Liao, X.; Li, D.; Zhang, L. The prognostic value of the proteasome activator subunit gene family in skin cutaneous melanoma. J. Cancer 2019, 10, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Gibney, G.; Kudchadkar, R.R.; Yu, B.; Cheng, P.; Martinez, A.J.; Kroeger, J.; Richards, A.; McCormick, L.; Moberg, V.; et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol. Res. 2016, 4, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.S.; Gibney, G.; Sullivan, R.J.; Sosman, J.A.; Slingluff, C.L., Jr.; Lawrence, D.P.; Logan, T.F.; Schuchter, L.M.; Nair, S.; Fecher, L.; et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016, 17, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF Inhibition Is Associated with Enhanced Melanoma Antigen Expression and a More Favorable Tumor Microenvironment in Patients with Metastatic Melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Huynh, S.; Mortier, L.; Dutriaux, C.; Maubec, E.; Boileau, M.; Dereure, O.; Leccia, M.-T.; Arnault, J.P.; Brunet-Possenti, F.; Aubin, F.; et al. Combined Therapy with Anti-PD1 and BRAF and/or MEK Inhibitor for Advanced Melanoma: A Multicenter Cohort Study. Cancers 2020, 12, 1666. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Hervieu, A.; Rébé, C.; Végran, F.; Chalmin, F.; Bruchard, M.; Vabres, P.; Apetoh, L.; Ghiringhelli, F.; Mignot, G. Dacarbazine-Mediated Upregulation of NKG2D Ligands on Tumor Cells Activates NK and CD8 T Cells and Restrains Melanoma Growth. J. Investig. Dermatol. 2013, 133, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugurel, S.; Paschen, A.; Becker, J.C. Dacarbazine in Melanoma: From a Chemotherapeutic Drug to an Immunomodulating Agent. J. Investig. Dermatol. 2013, 133, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.Y.; Li, J.; Tao, L.; Lam, A.K.; Chan, K.W.; Ko, J.M.Y.; Yu, V.Z.; Wong, M.; Li, B.; Lung, M.L. Chemotherapeutic Treatments Increase PD-L1 Expression in Esophageal Squamous Cell Carcinoma through EGFR/ERK Activation. Transl. Oncol. 2018, 11, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front. Immunol. 2018, 9, 1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadash-Bengad, R.; Hajaj, E.; Klein, S.; Merims, S.; Frank, S.; Eisenberg, G.; Yakobson, A.; Orevi, M.; Caplan, N.; Peretz, T.; et al. Immunotherapy Potentiates the Effect of Chemotherapy in Metastatic Melanoma—A Retrospective Study. Front. Oncol. 2020, 10, 70. [Google Scholar] [CrossRef]
- Vera Aguilera, J.; Paludo, J.; McWilliams, R.R.; Zhang, H.; Li, Y.; Kumar, A.B.; Failing, J.; Kottschade, L.A.; Block, M.S.; Markovic, S.N.; et al. Chemo-immunotherapy combination after PD-1 inhibitor failure improves clinical outcomes in metastatic melanoma patients. Melanoma Res. 2020, 30, 364–375. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Medrano, R.F.V.; Hunger, A.; Mendonça, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef] [Green Version]
- Davar, D.; Wang, H.; Chauvin, J.-M.; Pagliano, O.; Fourcade, J.J.; Ka, M.; Menna, C.; Rose, A.; Sander, C.; Borhani, A.A.; et al. Phase Ib/II Study of Pembrolizumab and Pegylated-Interferon Alfa-2b in Advanced Melanoma. J. Clin. Oncol. 2018, 36, 3450. [Google Scholar] [CrossRef]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of Cytokine-Induced Myeloid-Derived Suppressor Cells from Normal Human Peripheral Blood Mononuclear Cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Mundy-Bosse, B.L.; Lesinski, G.B.; Jaimeramirez, A.C.; Benninger, K.; Khan, M.; Kuppusamy, P.; Guenterberg, K.; Kondadasula, S.V.; Chaudhury, A.R.; La Perle, K.M.; et al. Myeloid-Derived Suppressor Cell Inhibition of the IFN Response in Tumor-Bearing Mice. Cancer Res. 2011, 71, 5101–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, J.; Wang, J.; VanGundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, S.K.; Ott, M.J.; Mostofa, A.G.M.; Chen, Z.; Chen, Y.A.; Kroeger, J.; Cao, B.; Mailloux, A.W.; Agrawal, A.; Schaible, B.J.; et al. Multi-Dimensional Flow Cytometry Analyses Reveal a Dichotomous Role for Nitric Oxide in Melanoma Patients Receiving Immunotherapy. Front. Immunol. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Riley, N.M.; Hebert, A.S.; Coon, J.J. Proteomics Moves into the Fast Lane. Cell Syst. 2016, 2, 142–143. [Google Scholar] [CrossRef] [Green Version]
- Kulak, N.A.; Geyer, P.E.; Mann, M. Loss-less Nano-fractionator for High Sensitivity, High Coverage Proteomics. Mol. Cell. Proteom. 2017, 16, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Severson, E.; Pignon, J.-C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Racle, J.; De Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, S.K.; Welsh, E.A.; Fang, B.; Hernandez, Y.I.; Rose, T.; Gray, J.; Koomen, J.M.; Berglund, A.; Mulé, J.J.; Markowitz, J. Multi-Omics and Informatics Analysis of FFPE Tissues Derived from Melanoma Patients with Long/Short Responses to Anti-PD1 Therapy Reveals Pathways of Response. Cancers 2020, 12, 3515. https://doi.org/10.3390/cancers12123515
Garg SK, Welsh EA, Fang B, Hernandez YI, Rose T, Gray J, Koomen JM, Berglund A, Mulé JJ, Markowitz J. Multi-Omics and Informatics Analysis of FFPE Tissues Derived from Melanoma Patients with Long/Short Responses to Anti-PD1 Therapy Reveals Pathways of Response. Cancers. 2020; 12(12):3515. https://doi.org/10.3390/cancers12123515
Chicago/Turabian StyleGarg, Saurabh K., Eric A. Welsh, Bin Fang, Yuliana I. Hernandez, Trevor Rose, Jhanelle Gray, John M. Koomen, Anders Berglund, James J. Mulé, and Joseph Markowitz. 2020. "Multi-Omics and Informatics Analysis of FFPE Tissues Derived from Melanoma Patients with Long/Short Responses to Anti-PD1 Therapy Reveals Pathways of Response" Cancers 12, no. 12: 3515. https://doi.org/10.3390/cancers12123515
APA StyleGarg, S. K., Welsh, E. A., Fang, B., Hernandez, Y. I., Rose, T., Gray, J., Koomen, J. M., Berglund, A., Mulé, J. J., & Markowitz, J. (2020). Multi-Omics and Informatics Analysis of FFPE Tissues Derived from Melanoma Patients with Long/Short Responses to Anti-PD1 Therapy Reveals Pathways of Response. Cancers, 12(12), 3515. https://doi.org/10.3390/cancers12123515