Synthetic MIR143-3p Suppresses Cell Growth in Rhabdomyosarcoma Cells by Interrupting RAS Pathways Including PAX3–FOXO1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
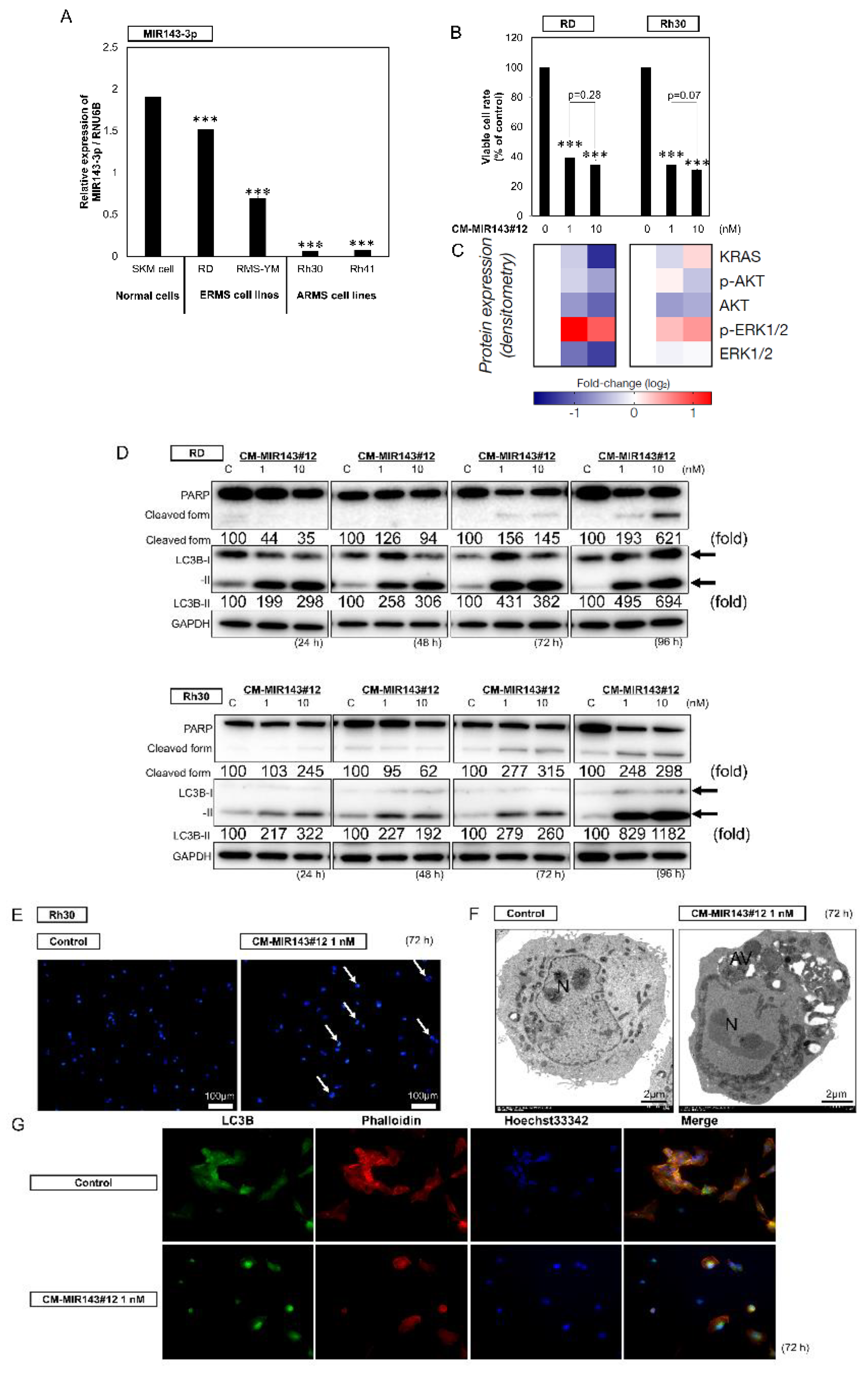
2.1. Downregulated Expression of MIR143-3p in RMS Cell Lines
2.2. Growth Inhibitory Effect of CM-MIR143#12 on RMS Cells
2.3. AKT Inhibitor Induced Apoptosis and Autophagy in RMS Cells
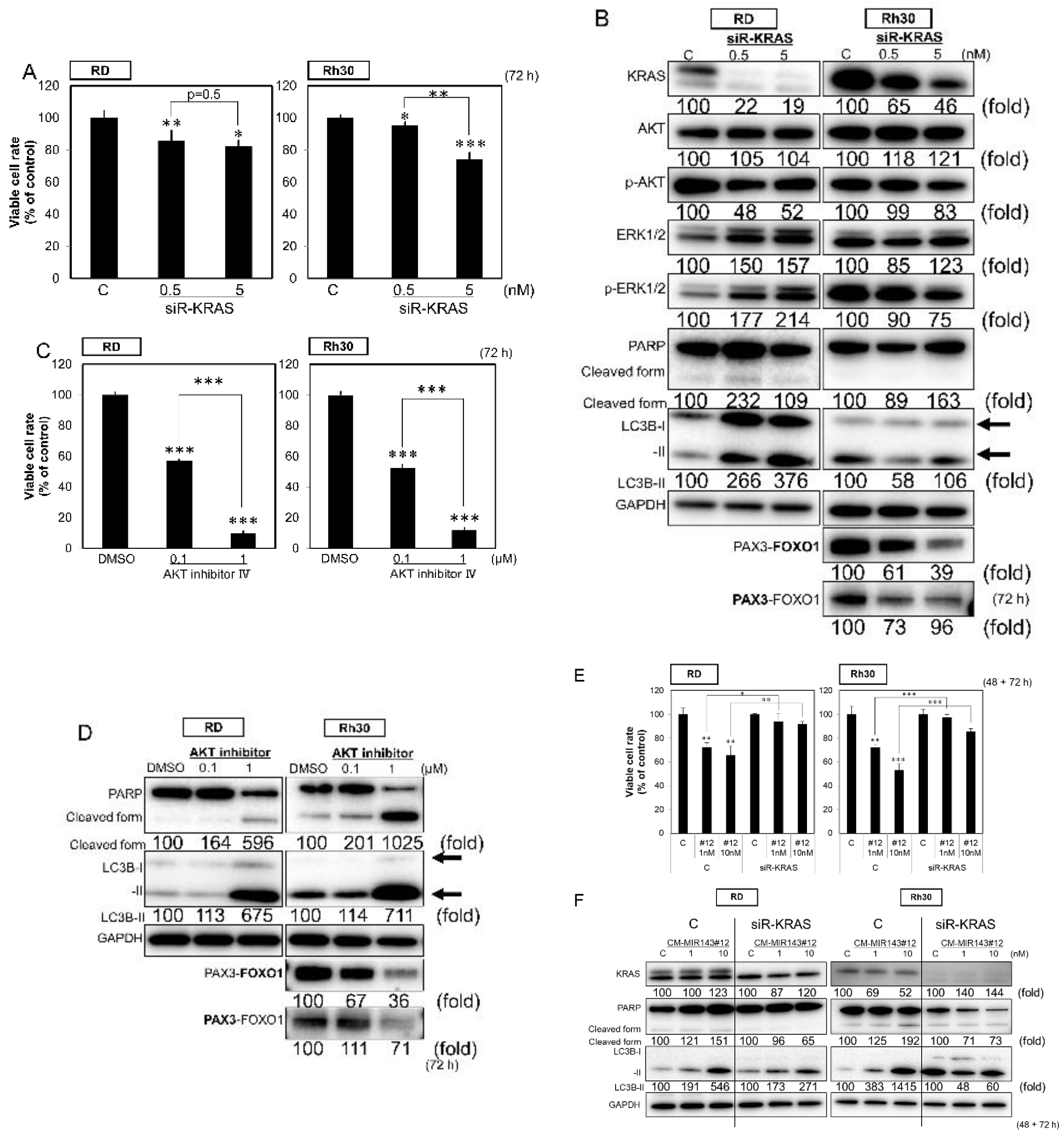
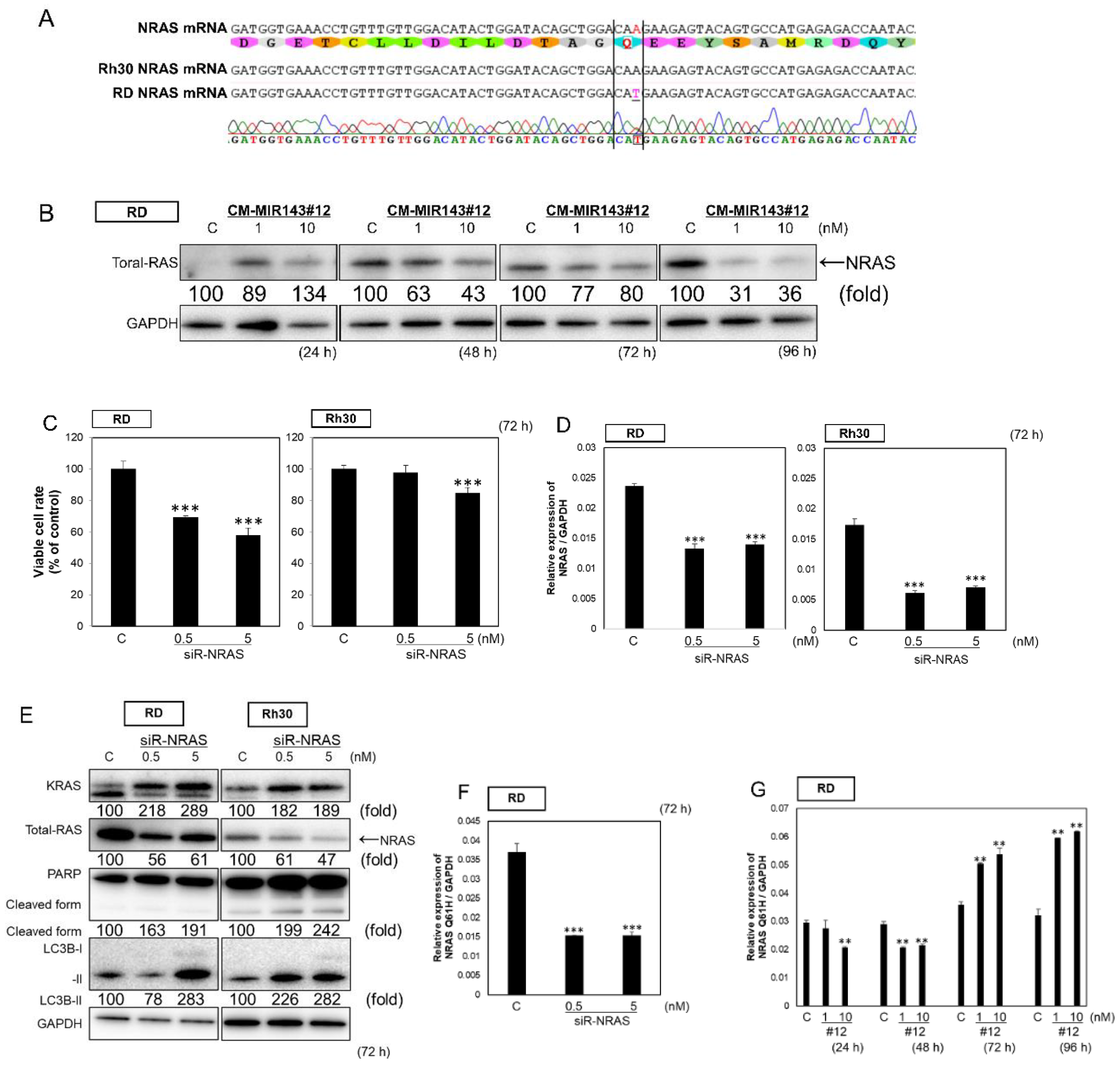
2.4. CM-MIR-143#12 Inhibits Cell Growth by Silencing NRAS in NRAS-Mutated RD Cell Lines
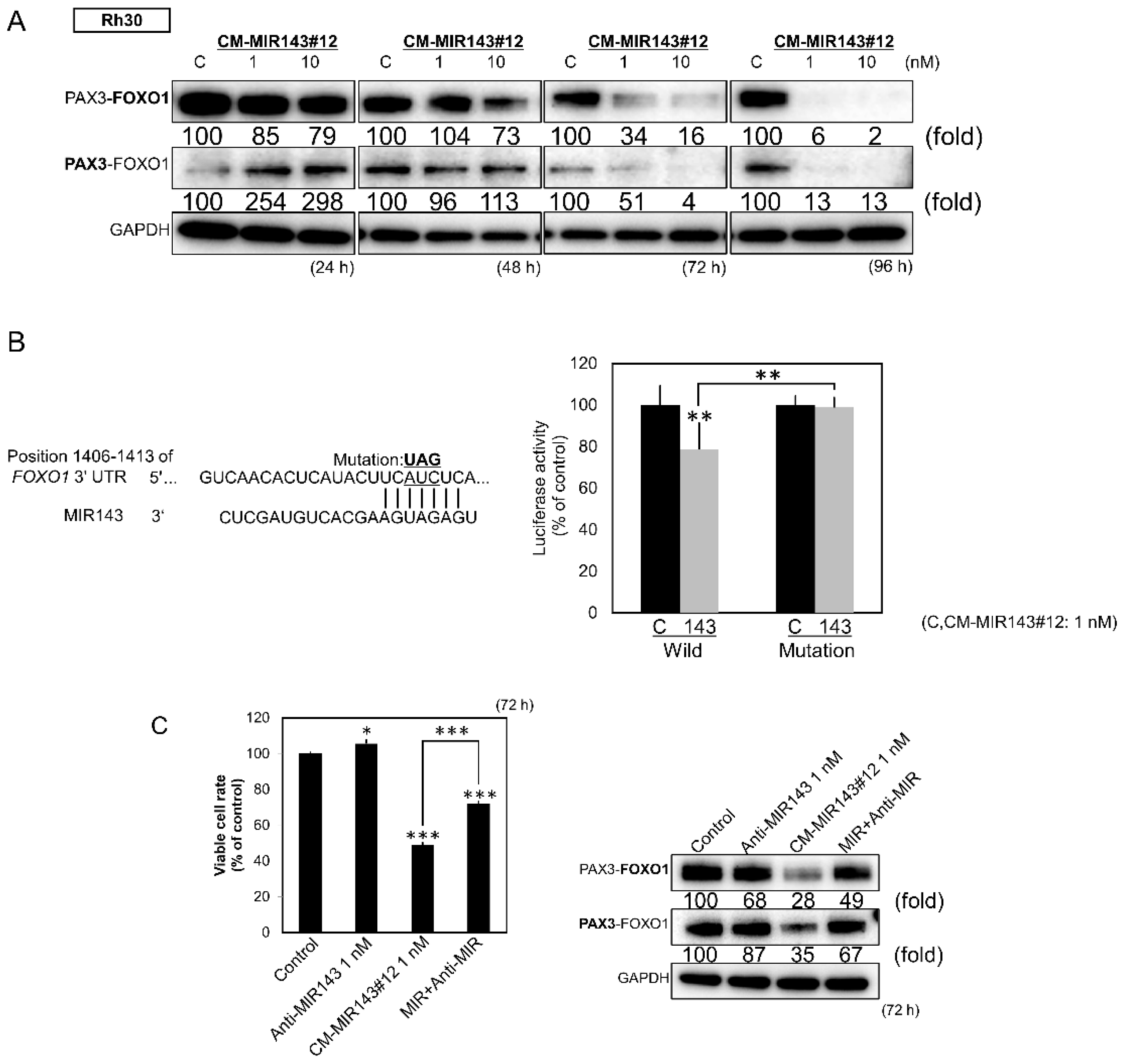
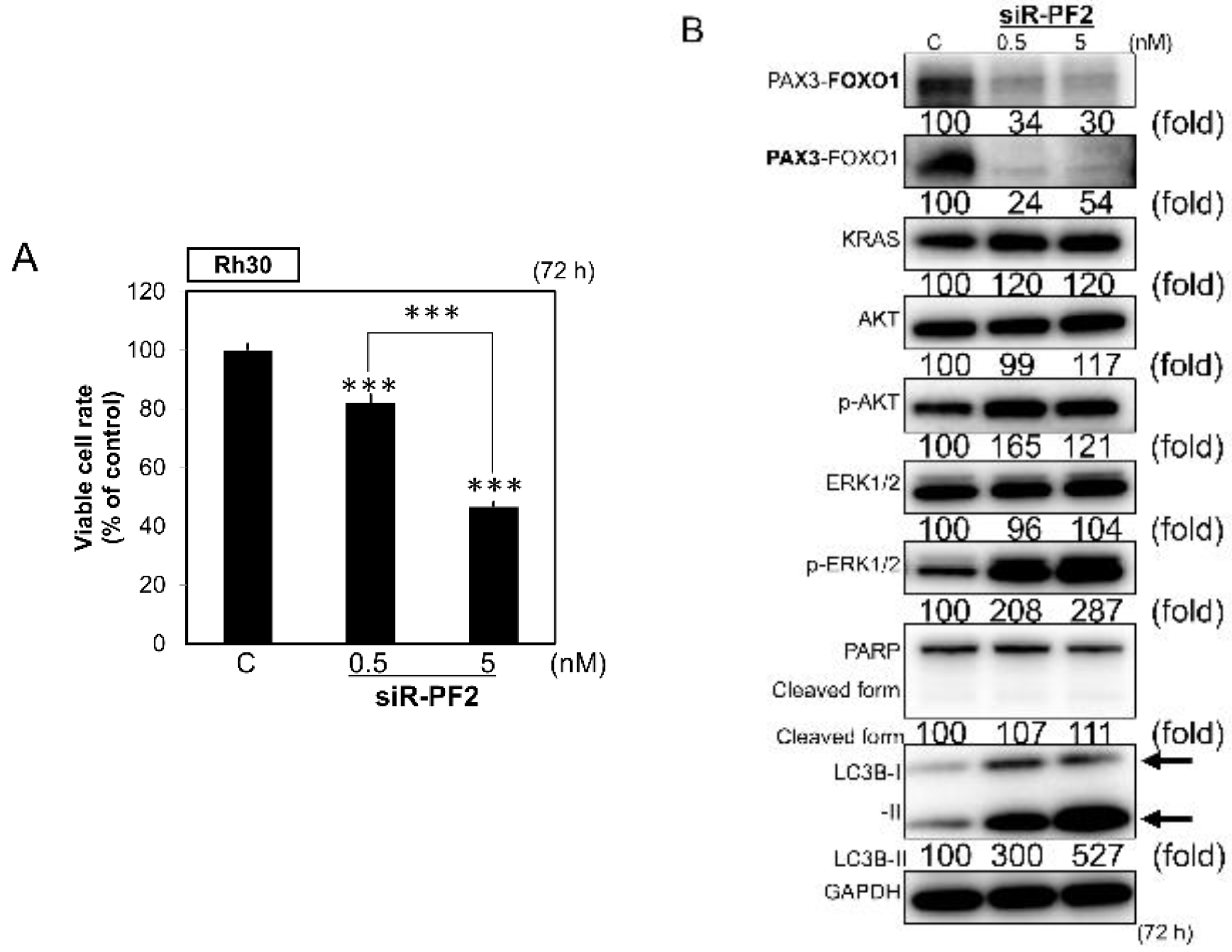
2.5. Direct Knockdown of the Pax3-Foxo1 Chimeric Gene By CM-MIR143#12 in Rh30 Cells
2.6. Knockdown of PAX3–FOXO1 Induced Autophagy in RMS Cell Lines
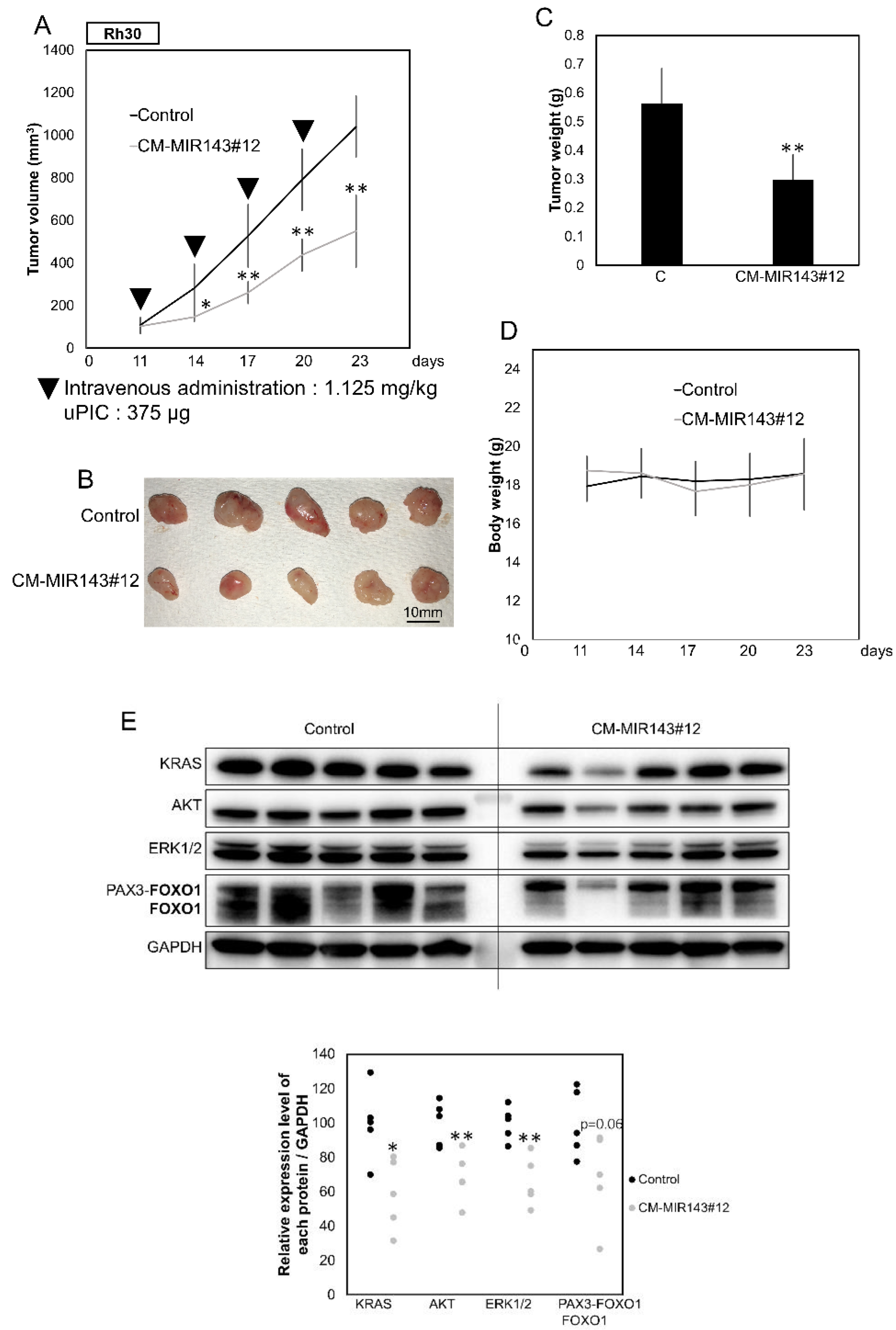
2.7. Tumor-Suppressing Effect of CM-MIR143#12 on Rh30 Cell-Xenografted Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Viability
4.2. Transfection Experiments
4.3. Inhibitor Agents
4.4. Western Blotting
4.5. Quantitative Real-Time PCR (qPCR)
4.6. Luciferase Reporter Assay
4.7. Hoechst 33342 Staining
4.8. Electron Microscopy
4.9. Immunofluorescence (IFC) Study
4.10. Human Tumor Xenograft Model
4.11. RNA Samples from Skeletal Muscle Cells
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J., 3rd; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, W.J.; Galili, N.; Mukhopadhyay, S.; Rovera, G.; Bennicelli, J.; Barr, F.G.; Rauscher, F.J., 3rd. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell. Biol. 1995, 15, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Shelton, G.D.; Cavenee, W.K.; Arden, K.C. Embryonic expression of the tumor-associated PAX3-FKHR fusion protein interferes with the developmental functions of Pax3. Proc. Natl. Acad. Sci. USA 2001, 98, 1589–1594. [Google Scholar] [CrossRef]
- Meza, J.L.; Anderson, J.; Pappo, A.S.; Meyer, W.H.; Children’s Oncology, G. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The children’s oncology group. J. Clin. Oncol. 2006, 24, 3844–3851. [Google Scholar] [CrossRef]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma—The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet Muscle 2012, 2, 25. [Google Scholar] [CrossRef]
- Arnold, M.A.; Barr, F.G. Molecular diagnostics in the management of rhabdomyosarcoma. Expert Rev. Mol. Diagn. 2017, 17, 189–194. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun. 2015, 6, 7557. [Google Scholar] [CrossRef]
- Crose, L.E.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef]
- Nakagawa, N.; Kikuchi, K.; Yagyu, S.; Miyachi, M.; Iehara, T.; Tajiri, T.; Sakai, T.; Hosoi, H. Mutations in the RAS pathway as potential precision medicine targets in treatment of rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2019, 512, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, H. Current status of treatment for pediatric rhabdomyosarcoma in the USA and Japan. Pediatr. Int. 2016, 58, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Dorado Garcia, H.; Scheer, M.; Henssen, A.G. Current and future treatment strategies for rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Ambros, V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell 2003, 113, 673–676. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Naoe, T. Let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Miyachi, M.; Tsuchiya, K.; Yoshida, H.; Yagyu, S.; Kikuchi, K.; Misawa, A.; Iehara, T.; Hosoi, H. Circulating muscle-specific microRNA, miR-206, as a potential diagnostic marker for rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2010, 400, 89–93. [Google Scholar] [CrossRef]
- Taulli, R.; Bersani, F.; Foglizzo, V.; Linari, A.; Vigna, E.; Ladanyi, M.; Tuschl, T.; Ponzetto, C. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J. Clin. Investig. 2009, 119, 2366–2378. [Google Scholar] [CrossRef]
- Hanna, J.A.; Garcia, M.R.; Go, J.C.; Finkelstein, D.; Kodali, K.; Pagala, V.; Wang, X.; Peng, J.; Hatley, M.E. PAX7 is a required target for microRNA-206-induced differentiation of fusion-negative rhabdomyosarcoma. Cell Death Dis. 2016, 7, e2256. [Google Scholar] [CrossRef]
- Sugito, N.; Taniguchi, K.; Kuranaga, Y.; Ohishi, M.; Soga, T.; Ito, Y.; Miyachi, M.; Kikuchi, K.; Hosoi, H.; Akao, Y. Cancer-specific energy metabolism in rhabdomyosarcoma cells is regulated by microRNA. Nucleic Acid Ther. 2017, 27, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Iwasaki, J.; Kumazaki, M.; Mori, T.; Maruo, K.; Sakai, H.; Yamada, N.; Shimada, K.; Naoe, T.; Kitade, Y.; et al. Chemically modified synthetic microRNA-205 inhibits the growth of melanoma cells in vitro and in vivo. Mol. Ther. 2013, 21, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Ito, Y.; Otsuki, Y.; Uno, B.; Uchiyama, K.; et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett. 2015, 363, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Inamoto, T.; Takahara, K.; Takai, T.; Yoshikawa, Y.; Kiyama, S.; Akao, Y.; et al. MiR-145 negatively regulates Warburg effect by silencing KLF4 and PTBP1 in bladder cancer cells. Oncotarget 2017, 8, 33064–33077. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Kitade, Y.; Kinoshita, T.; Naoe, T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007, 98, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Maruo, K.; Yamada, N.; Kitade, Y.; Naoe, T.; Akao, Y. MicroRNA-143 functions as a tumor suppressor in human bladder cancer T24 cells. Cancer Lett. 2011, 307, 211–220. [Google Scholar] [CrossRef]
- Ahmad, I.; Singh, L.B.; Yang, Z.H.; Kalna, G.; Fleming, J.; Fisher, G.; Cooper, C.; Cuzick, J.; Berney, D.M.; Moller, H.; et al. Mir143 expression inversely correlates with nuclear ERK5 immunoreactivity in clinical prostate cancer. Br. J. Cancer 2013, 108, 149–154. [Google Scholar] [CrossRef]
- Cheng, T.; Hu, C.; Yang, H.; Cao, L.; An, J. Transforming growth factor-beta-induced miR143 expression in regulation of non-small cell lung cancer cell viability and invasion capacity in vitro and in vivo. Int. J. Oncol. 2014, 45, 1977–1988. [Google Scholar] [CrossRef]
- Shen, J.Z.; Zhang, Y.Y.; Fu, H.Y.; Wu, D.S.; Zhou, H.R. Overexpression of microRNA-143 inhibits growth and induces apoptosis in human leukemia cells. Oncol. Rep. 2014, 31, 2035–2042. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Liu, H.; Wang, H.; Liu, H.; Chen, Y. Effect of miR-143 on the apoptosis of osteosarcoma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 14241–14246. [Google Scholar] [PubMed]
- Li, W.H.; Wu, H.J.; Li, Y.X.; Pan, H.G.; Meng, T.; Wang, X. MicroRNA-143 promotes apoptosis of osteosarcoma cells by caspase-3 activation via targeting Bcl-2. Biomed. Pharmacother. 2016, 80, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Dai, G.; Yu, L.; Hu, Q.; Chen, J.; Guo, W. miR-143-3p inhibits the proliferation, migration and invasion in osteosarcoma by targeting FOSL2. Sci. Rep. 2018, 8, 606. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Feng, H.; Jiao, J.; Qian, L.; Sun, B.; Chen, P.; Li, Q.; Liang, Z. Mechanism of miR-143-3p inhibiting proliferation, migration and invasion of osteosarcoma cells by targeting MAPK7. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2065–2071. [Google Scholar] [CrossRef]
- Akao, Y.; Kumazaki, M.; Shinohara, H.; Sugito, N.; Kuranaga, Y.; Tsujino, T.; Yoshikawa, Y.; Kitade, Y. Impairment of K-Ras signaling networks and increased efficacy of epidermal growth factor receptor inhibitors by a novel synthetic miR-143. Cancer Sci. 2018, 109, 1455–1467. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Taniguchi, K.; Tsujino, T.; Heishima, K.; Inamoto, T.; Takai, T.; Minami, K.; Azuma, H.; Miyata, K.; Hayashi, K.; et al. Anti-cancer effects of a chemically modified miR-143 on Bladder cancer by either systemic or intravesical treatment. Mol. Ther. Methods Clin. Dev. 2019, 13, 290–302. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Tajirika, T.; Sugito, N.; Kuranaga, Y.; Shinohara, H.; Tsujino, T.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K. Synthetic miR-143 inhibits growth of HER2-positive gastric cancer cells by suppressing KRAS networks including DDX6 RNA helicase. Int. J. Mol. Sci. 2019, 20, 1697. [Google Scholar] [CrossRef]
- Felix, C.A.; Kappel, C.C.; Mitsudomi, T.; Nau, M.M.; Tsokos, M.; Crouch, G.D.; Nisen, P.D.; Winick, N.J.; Helman, L.J. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 1992, 52, 2243–2247. [Google Scholar]
- Liu, X.; Zhang, Y.; Wang, S.; Liu, G.; Ruan, L. Loss of miR-143 and miR-145 in condyloma acuminatum promotes cellular proliferation and inhibits apoptosis by targeting NRAS. R Soc. Open Sci. 2018, 5, 172376. [Google Scholar] [CrossRef]
- Kikuchi, K.; Tsuchiya, K.; Otabe, O.; Gotoh, T.; Tamura, S.; Katsumi, Y.; Yagyu, S.; Tsubai-Shimizu, S.; Miyachi, M.; Iehara, T.; et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2008, 365, 568–574. [Google Scholar] [CrossRef]
- Dolgikh, N.; Hugle, M.; Vogler, M.; Fulda, S. NRAS-mutated rhabdomyosarcoma cells are vulnerable to mitochondrial apoptosis induced by coinhibition of MEK and PI3Kalpha. Cancer Res. 2018, 78, 2000–2013. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Iio, A.; Naoe, T. Role of microRNA-143 in Fas-mediated apoptosis in human T-cell leukemia Jurkat cells. Leuk. Res. 2009, 33, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Yamada, N.; Naito, S.; Akao, Y. Replacement treatment with microRNA-143 and -145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 2013, 328, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.B.; Soon, Y.L.; Nguyen, P.N.; Hiew, M.S.; Huang, C.J. MicroRNA-5p and -3p co-expression and cross-targeting in colon cancer cells. J. Biomed. Sci. 2014, 21, 95. [Google Scholar] [CrossRef]
- Zhao, W.G.; Yu, S.N.; Lu, Z.H.; Ma, Y.H.; Gu, Y.M.; Chen, J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis 2010, 31, 1726–1733. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, S.; Chen, Y.; Zhao, J.; Zhang, K.; Wang, J.; Chen, S. microRNA-143 is associated with the survival of ALDH1+CD133+ osteosarcoma cells and the chemoresistance of osteosarcoma. Exp. Biol. Med. (Maywood) 2015, 240, 867–875. [Google Scholar] [CrossRef]
- Lin, X.T.; Zheng, X.B.; Fan, D.J.; Yao, Q.Q.; Hu, J.C.; Lian, L.; Wu, X.J.; Lan, P.; He, X.S. MicroRNA-143 targets ATG2B to inhibit autophagy and increase inflammatory responses in Crohn’s disease. Inflamm. Bowel Dis. 2018, 24, 781–791. [Google Scholar] [CrossRef]
- Tsujino, T.; Sugito, N.; Taniguchi, K.; Honda, R.; Komura, K.; Yoshikawa, Y.; Takai, T.; Minami, K.; Kuranaga, Y.; Shinohara, H.; et al. MicroRNA-143/Musashi-2/KRAS cascade contributes positively to carcinogenesis in human bladder cancer. Cancer Sci. 2019, 110, 2189–2199. [Google Scholar] [CrossRef]
- Gokita, K.; Inoue, J.; Ishihara, H.; Kojima, K.; Inazawa, J. Therapeutic potential of LNP-mediated delivery of miR-634 for cancer therapy. Mol. Ther. Nucleic Acids 2019, 19, 330–338. [Google Scholar] [CrossRef]
- Watanabe, S.; Hayashi, K.; Toh, K.; Kim, H.J.; Liu, X.; Chaya, H.; Fukushima, S.; Katsushima, K.; Kondo, Y.; Uchida, S.; et al. In vivo rendezvous of small nucleic acid drugs with charge-matched block catiomers to target cancers. Nat. Commun. 2019, 10, 1894. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ishihara, H. Structure, activity and uptake mechanism of siRNA-lipid nanoparticles with an asymmetric ionizable lipid. Int. J. Pharm. 2016, 510, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Gary, D.J.; Puri, N.; Won, Y.Y. Polymer-based siRNA delivery: Perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J. Control. Release 2007, 121, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Target Scan Human. Prediction of microRNA Targets. Available online: http://www.targetscan.org/ (accessed on 9 November 2020).
- Tang, Y.; Simoneau, A.R.; Liao, W.X.; Yi, G.; Hope, C.; Liu, F.; Li, S.; Xie, J.; Holcombe, R.F.; Jurnak, F.A.; et al. WIF1, a Wnt pathway inhibitor, regulates SKP2 and c-myc expression leading to G1 arrest and growth inhibition of human invasive urinary bladder cancer cells. Mol. Cancer Ther. 2009, 8, 458–468. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugito, N.; Heishima, K.; Ito, Y.; Akao, Y. Synthetic MIR143-3p Suppresses Cell Growth in Rhabdomyosarcoma Cells by Interrupting RAS Pathways Including PAX3–FOXO1. Cancers 2020, 12, 3312. https://doi.org/10.3390/cancers12113312
Sugito N, Heishima K, Ito Y, Akao Y. Synthetic MIR143-3p Suppresses Cell Growth in Rhabdomyosarcoma Cells by Interrupting RAS Pathways Including PAX3–FOXO1. Cancers. 2020; 12(11):3312. https://doi.org/10.3390/cancers12113312
Chicago/Turabian StyleSugito, Nobuhiko, Kazuki Heishima, Yuko Ito, and Yukihiro Akao. 2020. "Synthetic MIR143-3p Suppresses Cell Growth in Rhabdomyosarcoma Cells by Interrupting RAS Pathways Including PAX3–FOXO1" Cancers 12, no. 11: 3312. https://doi.org/10.3390/cancers12113312
APA StyleSugito, N., Heishima, K., Ito, Y., & Akao, Y. (2020). Synthetic MIR143-3p Suppresses Cell Growth in Rhabdomyosarcoma Cells by Interrupting RAS Pathways Including PAX3–FOXO1. Cancers, 12(11), 3312. https://doi.org/10.3390/cancers12113312