Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. MF Achieves Low µM Serum Concentrations in Inhα/Tag Mouse Serum
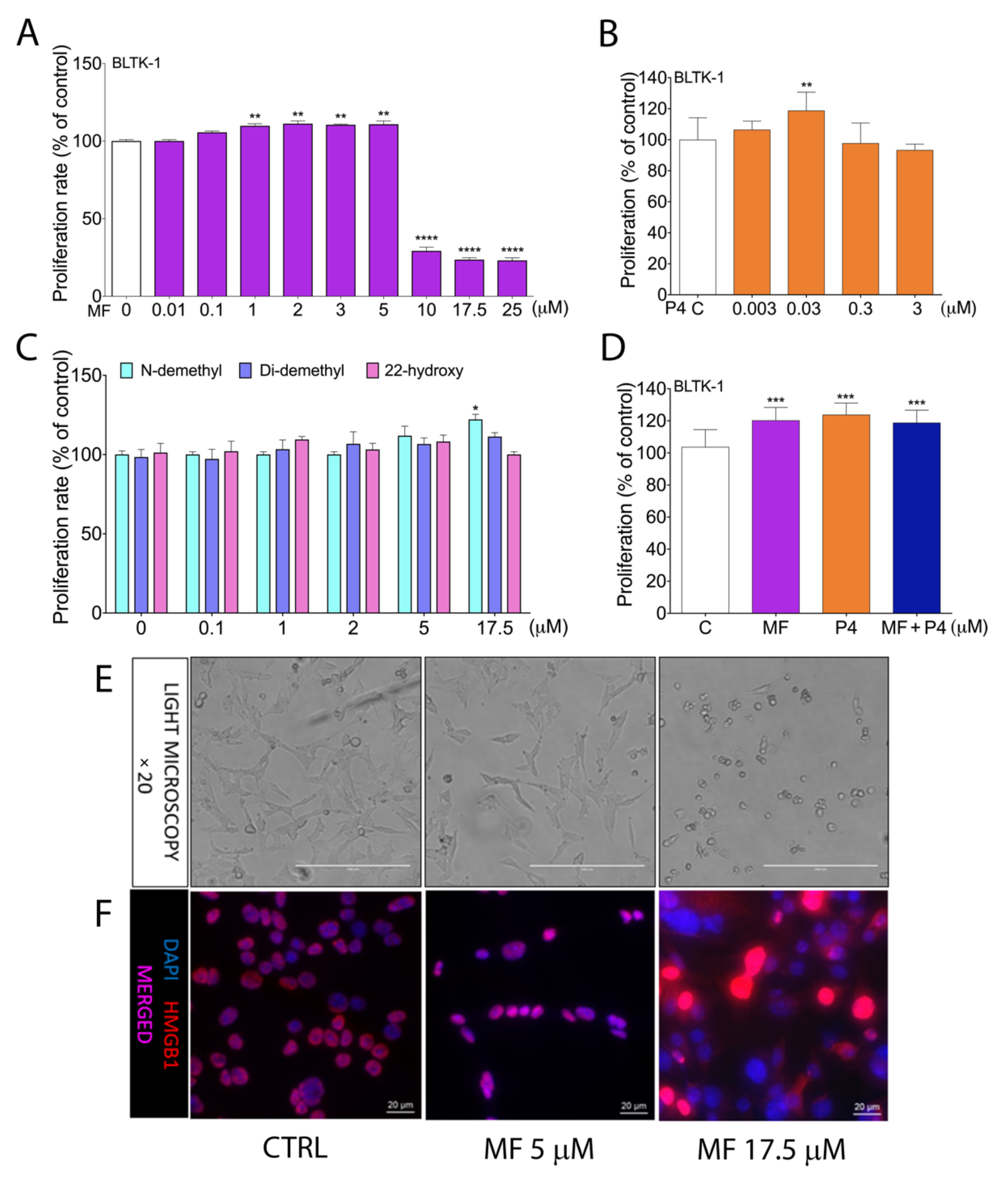
2.2. MF and P4 Treatmenst Affect Leydig Tumor Cell Proliferation In Vitro
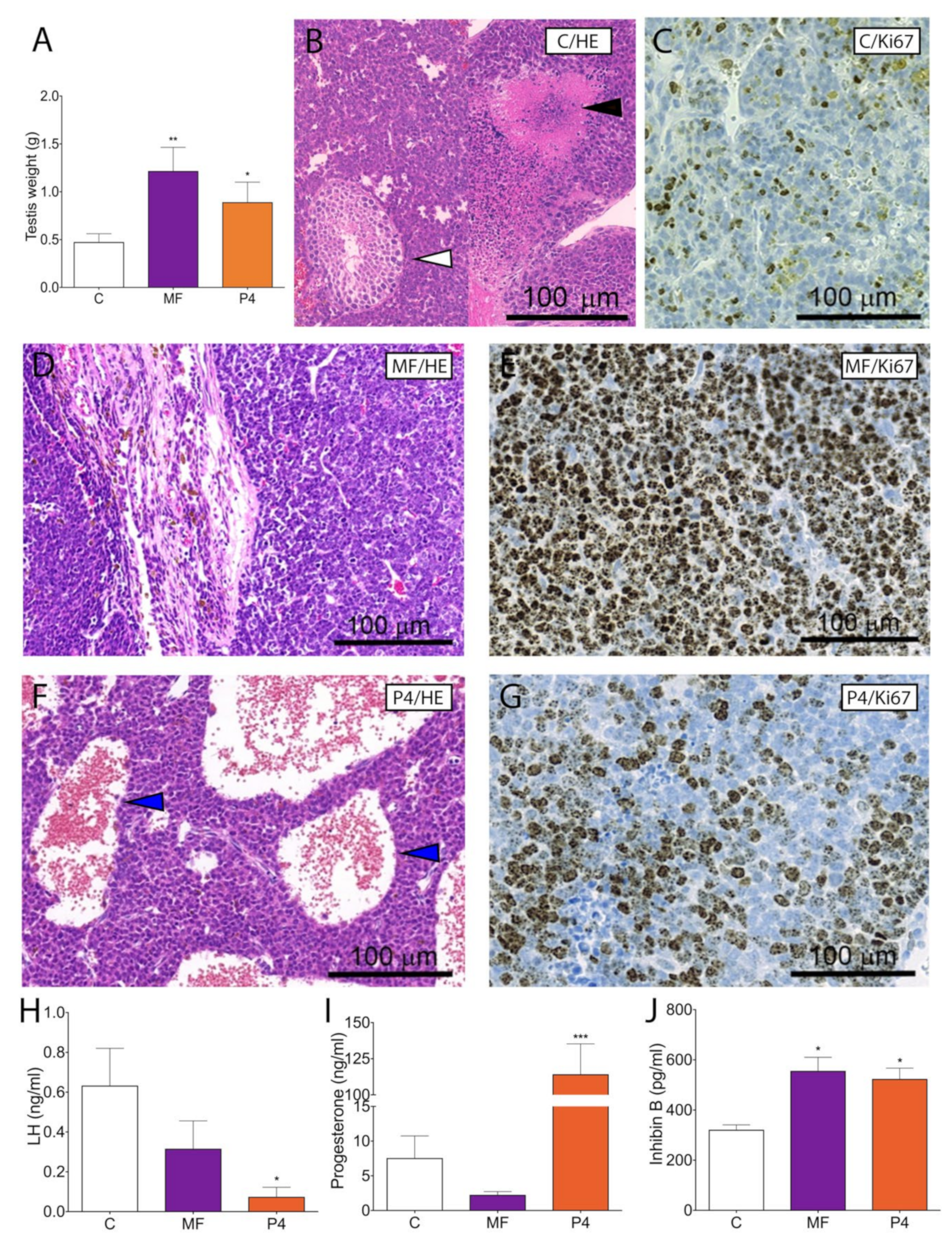
2.3. MF and P4 Stimulate Leydig Cell Tumor Growth In Vivo
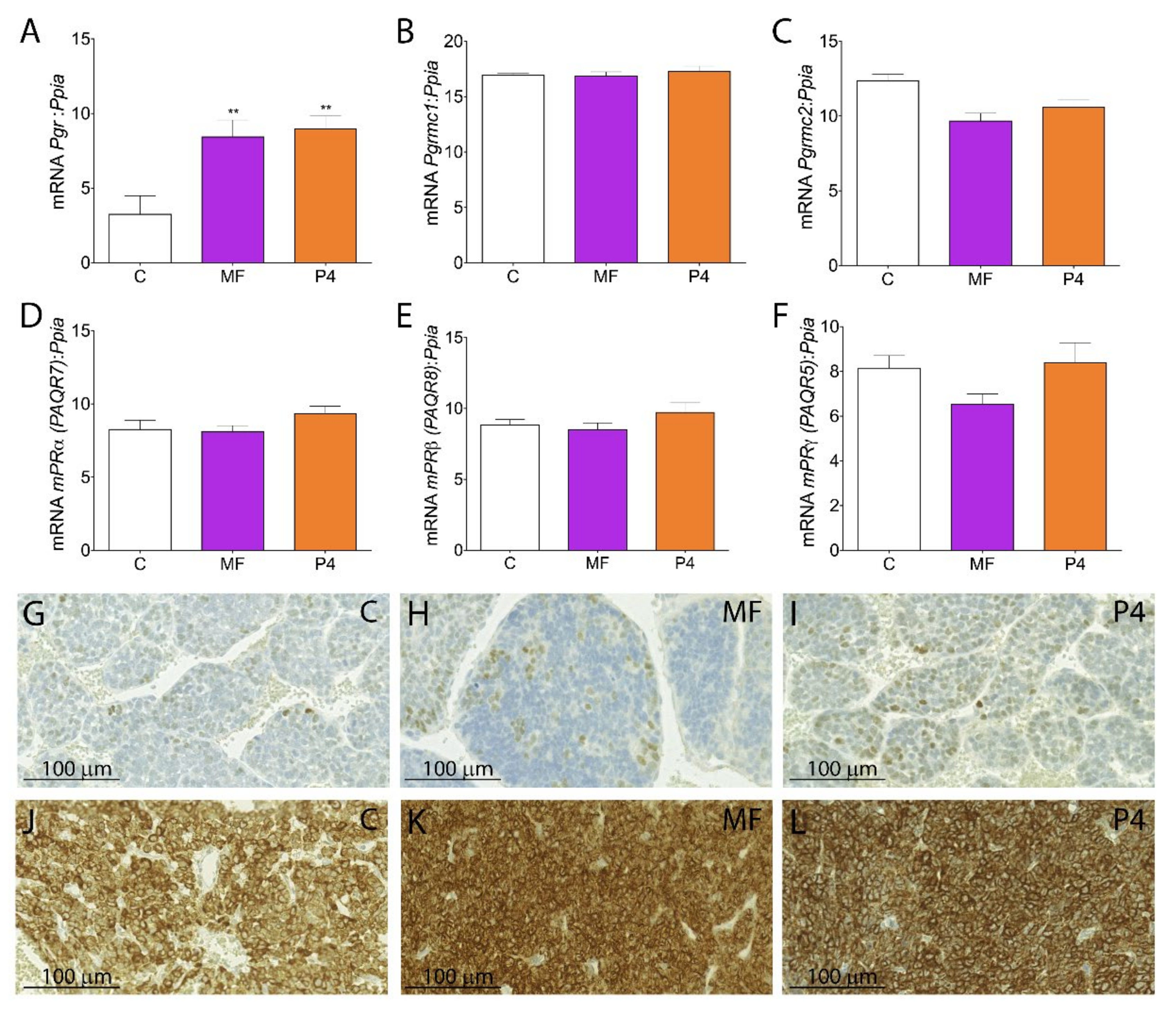
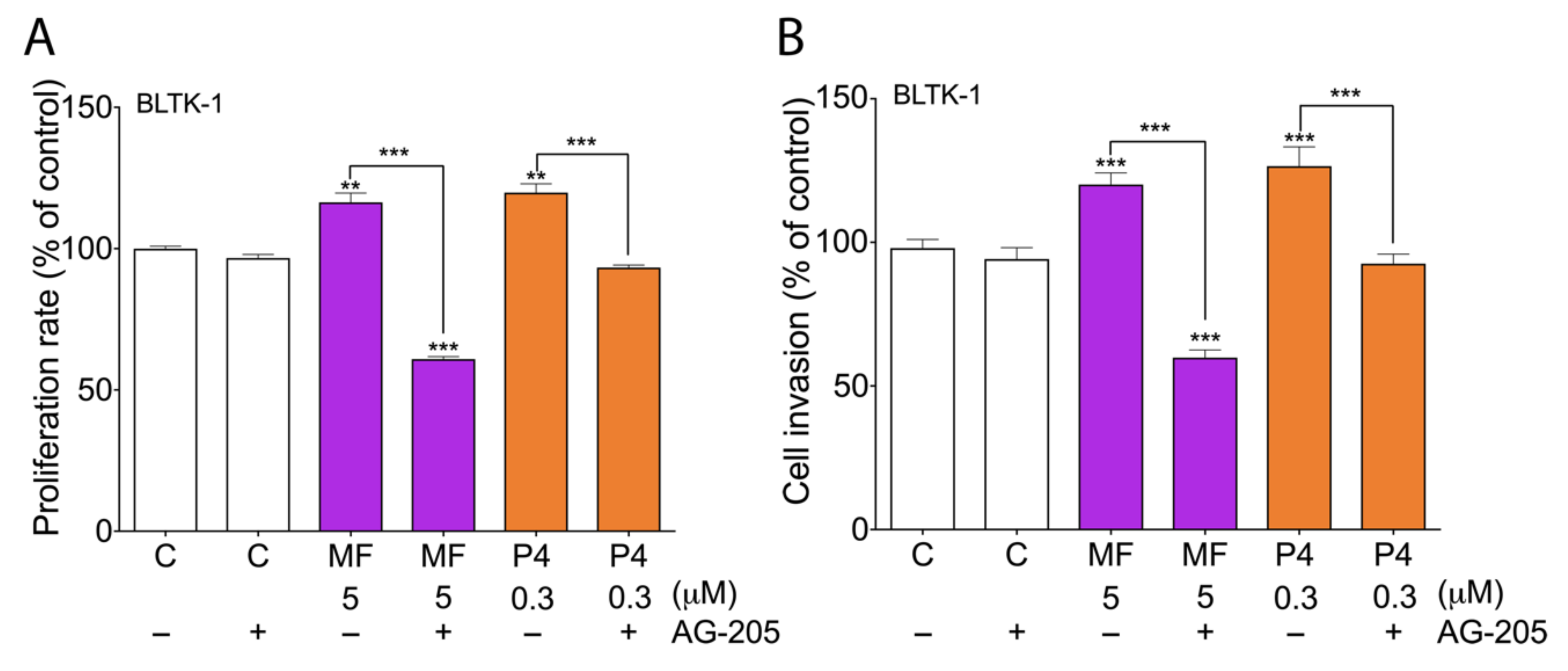
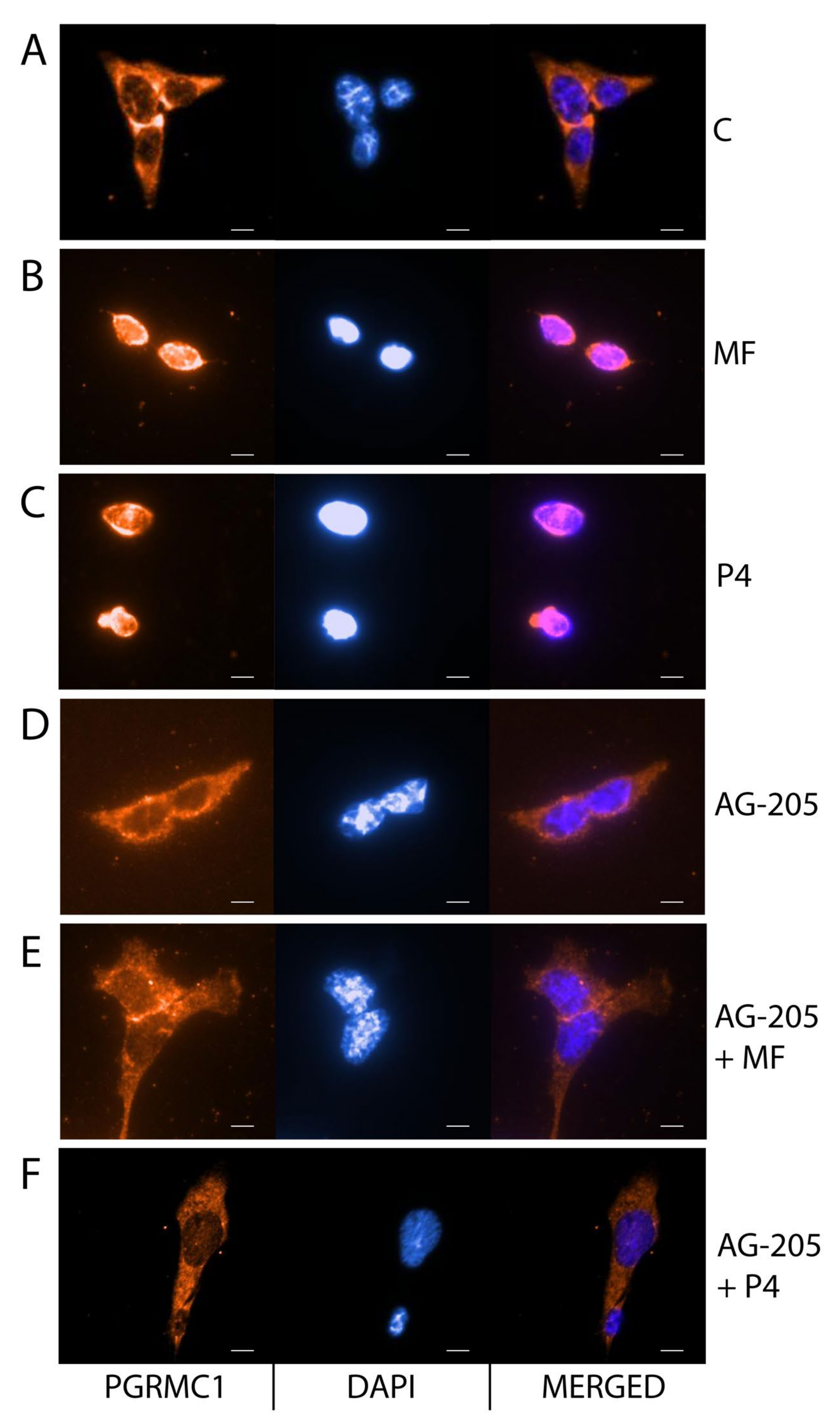
2.4. MF and P4 Stimulate Leydig Cell Tumors Proliferation and Invasiveness through Pgrmc1, Independently of Their Glucocorticoid Receptors
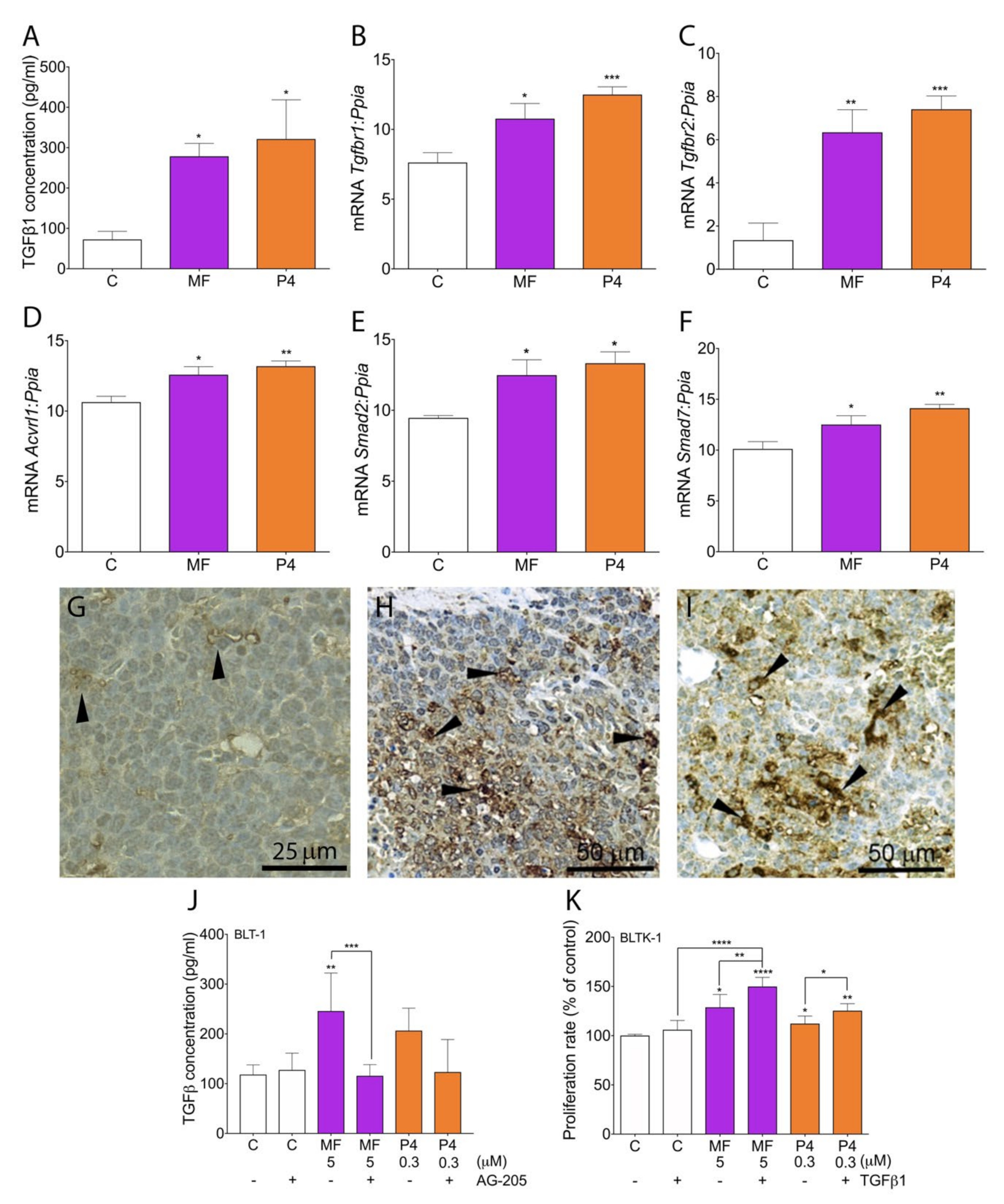
2.5. MF and P4 Promote Tumor Progression through Activation of the TGFβ1 Superfamily Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. MF Pharmacokinetics
4.3. Cell Cultures
4.4. Cell Proliferation
4.5. Cell Invasion
4.6. Histological and Immunohistochemical Analyses
4.7. Immunocytochemistry Analysis
4.8. Real Time RT-PCR
4.9. Hormones and TGFβ1 Measurement
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 22-hydroxy MF | (11β,17β)-11-[4-(Dimethylamino)phenyl]-17-hydroxy-17-(3-hydroxy-1-propyn-1-yl)-estra-4,9-dien-3-one |
| AG-205 | inhibitor PGRMC1 |
| BLTK-1 | Immortalized cell line from Inhα/Tag TG mice |
| DXM | Dexamethasone |
| Di-demethyl MF | (11β,17β)-11-(4-Aminophenyl)-17-hydroxy-17-(1-propyn-1-yl)-estra-4,9-dien-3-one |
| GR | Glucocorticoid receptor |
| HMGB1 | High mobility group box 1 protein |
| HSP90i | Glucocorticoid receptor inhibitor |
| Inhα/Tag | Transgenic mice expressing Simian Virus 40 T antigen under inhibin-α promoter |
| LC | Leydig cell |
| LCT | Leydig cell tumor |
| LHR | Luteinizing hormone receptor |
| MF | Mifepristone |
| mPR | Membrane progesterone receptor |
| N-demethyl MF | 11β,17β)-17-Hydroxy-11-[4-(methylamino)phenyl]-17-(1-propyn-1-yl)-estra-4,9-dien-3-one |
| P4 | Progesterone |
| PGR | Nuclear progesterone receptors |
| PGRMC1 | Progesterone receptor membrane component 1 |
| PR | Progesterone receptors |
| SPRM | Selective progesterone receptor modulator |
| StAR | Steroidogenic acute regulatory protein |
| TG | Transgenic |
| TGF-β1 | Transforming growth factor β1 |
References
- Meyer, M.E.; Pornon, A.; Ji, J.W.; Bocquel, M.T.; Chambon, P.; Gronemeyer, H. Agonistic and antagonistic activities of RU486 on the functions of the human progesterone receptor. EMBO J. 1990, 9, 3923–3932. [Google Scholar] [CrossRef]
- Perrault, D.; Eisenhauer, E.A.; Pritchard, K.I.; Panasci, L.; Norris, B.; Vandenberg, T.; Fisher, B. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcinoma: A National Cancer Institute of Canada Clinical Trials Group study. J. Clin. Oncol. 1996, 14, 2709–2712. [Google Scholar] [CrossRef] [PubMed]
- Rocereto, T.F.; Saul, H.M.; Aikins, J.A., Jr.; Paulson, J. Phase II study of mifepristone (RU486) in refractory ovarian cancer. Gynecol. Oncol. 2000, 77, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Ng, E.H.Y.; Tang, O.S. Mifepristone: Contraceptive and noncontraceptive uses. Curr. Opin. Obstet. Gynecol. 2002, 14, 325. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, S.M.; Weiss, M.H.; Russell, C.A.; Spitz, I.M.; Ahmadi, J.; Sadun, A.; Sitruk-Ware, R. Long-term administration of mifepristone (RU486): Clinical tolerance during extended treatment of meningioma. Cancer Investig. 2006, 24, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Ramondetta, L.M.; Johnson, A.J.; Sun, C.C.; Atkinson, N.; Smith, J.A.; Jung, M.S.; Broaddus, R.; Iyer, R.B.; Burke, T. Phase 2 trial of mifepristone (RU-486) in advanced or recurrent endometrioid adenocarcinoma or low-grade endometrial stromal sarcoma. Cancer 2009, 115, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Rocereto, T.F.; Brady, W.E.; Shahin, M.S.; Hoffman, J.S.; Small, L.; Rotmensch, J.; Mannel, R.S. A phase II evaluation of mifepristone in the treatment of recurrent or persistent epithelial ovarian, fallopian or primary peritoneal cancer: A gynecologic oncology group study. Gynecol. Oncol. 2010, 116, 332–334. [Google Scholar] [CrossRef]
- Tieszen, C.R.; Goyeneche, A.A.; Brandhagen, B.N.; Ortbahn, C.T.; Telleria, C.M. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer 2011, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Peluso, J.J. Multiplicity of progesterone’s actions and receptors in the mammalian ovary. Biol. Reprod. 2006, 75, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Goyeneche, A.A.; Caron, R.W.; Telleria, C.M. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin. Cancer Res. 2007, 13, 3370–3379. [Google Scholar] [CrossRef] [PubMed]
- Freeburg, E.M.; Goyeneche, A.A.; Seidel, E.E.; Telleria, C.M. Resistance to cisplatin does not affect sensitivity of human ovarian cancer cell lines to mifepristone cytotoxicity. Cancer Cell Int. 2009, 9, 4. [Google Scholar] [CrossRef]
- Gamarra-Luques, C.D.; Goyeneche, A.A.; Hapon, M.B.; Telleria, C.M. Mifepristone prevents repopulation of ovarian cancer cells escaping cisplatin-paclitaxel therapy. BMC Cancer 2012, 12, 200. [Google Scholar] [CrossRef] [PubMed]
- Ponikwicka-Tyszko, D.; Chrusciel, M.; Stelmaszewska, J.; Bernaczyk, P.; Chrusciel, P.; Sztachelska, M.; Scheinin, M.; Bidzinski, M.; Szamatowicz, J.; Huhtaniemi, I.T.; et al. Molecular mechanisms underlying mifepristone’s agonistic action on ovarian cancer progression. EBioMedicine 2019, 47, 170–183. [Google Scholar] [CrossRef]
- Kinkade, S. Testicular cancer. Am. Fam. Phys. 1999, 59, 2539–2544, 2549–2550. [Google Scholar] [PubMed]
- Rich, M.A.; Keating, M.A. Leydig cell tumors and tumors associated with congenital adrenal hyperplasia. Urol. Clin. N. Am. 2000, 27, 519–528. [Google Scholar] [CrossRef]
- Thomas, J.C.; Ross, J.H.; Kay, R. Stromal testis tumors in children: A report from the prepubertal testis tumor registry. J. Urol. 2001, 166, 2338–2340. [Google Scholar] [CrossRef]
- Hawkins, C.; Miaskowski, C. Testicular cancer: A review. Oncol. Nurs. Forum 1996, 23, 1203–1211; quiz 1212–1213. [Google Scholar]
- Pozza, C.; Pofi, R.; Tenuta, M.; Tarsitano, M.G.; Sbardella, E.; Fattorini, G.; Cantisani, V.; Lenzi, A.; Isidori, A.M.; Gianfrilli, D.; et al. Clinical presentation, management and follow-up of 83 patients with Leydig cell tumors of the testis: A prospective case-cohort study. Hum. Reprod. 2019, 34, 1389–1403. [Google Scholar] [CrossRef]
- McCluggage, W.G.; Shanks, J.H.; Arthur, K.; Banerjee, S.S. Cellular proliferation and nuclear ploidy assessments augment established prognostic factors in predicting malignancy in testicular Leydig cell tumours. Histopathology 1998, 33, 361–368. [Google Scholar] [CrossRef]
- Mostofi, F.K. Proceedings: Testicular tumors. Epidemiologic, etiologic, and pathologic features. Cancer 1973, 32, 1186–1201. [Google Scholar] [CrossRef]
- Al-Agha, O.M.; Axiotis, C.A. An in-depth look at Leydig cell tumor of the testis. Arch. Pathol. Lab. Med. 2007, 131, 311–317. [Google Scholar] [CrossRef]
- Gadkar-Sable, S.; Shah, C.; Rosario, G.; Sachdeva, G.; Puri, C. Progesterone receptors: Various forms and functions in reproductive tissues. Front. Biosci. 2005, 10, 2118–2130. [Google Scholar] [CrossRef]
- Pino, A.M.; Valladares, L.E. Evidence for a Leydig cell progesterone receptor in the rat. J. Steroid Biochem. 1988, 29, 709–714. [Google Scholar] [CrossRef]
- El-Hefnawy, T.; Huhtaniemi, I. Progesterone can participate in down-regulation of the luteinizing hormone receptor gene expression and function in cultured murine Leydig cells. Mol. Cell Endocrinol. 1998, 137, 127–138. [Google Scholar] [CrossRef]
- Due, W.; Dieckmann, K.P.; Loy, V.; Stein, H. Immunohistological determination of oestrogen receptor, progesterone receptor, and intermediate filaments in Leydig cell tumours, Leydig cell hyperplasia, and normal Leydig cells of the human testis. J. Pathol. 1989, 157, 225–234. [Google Scholar] [CrossRef]
- Shah, C.; Modi, D.; Sachdeva, G.; Gadkar, S.; Puri, C. Coexistence of intracellular and membrane-bound progesterone receptors in human testis. J. Clin. Endocrinol. Metab. 2005, 90, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Gonzaález, C.R.; Inserra, P.I.F.; Terradas, C.; Ponzio, R.; Puigdomenech, E.; Levalle, O.; Vitullo, A.D.; Calandra, R.S.; Gonzalez-Calvar, S.I. Altered Expression of Aromatase, Estrogen Receptors and Progesterone Receptors in Human Leydig Cell Hyperplasia. Adv. Biosci. Biotechnol. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Gonzalez, C.R.; Gonzalez, B.; Rulli, S.B.; Dos Santos, M.L.; Mattos Jardim Costa, G.; Franca, L.R.; Calandra, R.S.; Gonzalez-Calvar, S.I. TGF-beta1 system in Leydig cells. Part II: TGF-beta1 and progesterone, through Smad1/5, are involved in the hyperplasia/hypertrophy of Leydig cells. J. Reprod. Dev. 2010, 56, 400–404. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Manna, P.R.; Stocco, D.M.; Chakrabarti, G.; Mukhopadhyay, A.K. Stimulatory effect of progesterone on the expression of steroidogenic acute regulatory protein in MA-10 Leydig cells. Biol. Reprod. 2003, 68, 1054–1063. [Google Scholar] [CrossRef]
- El-Hefnawy, T.; Manna, P.R.; Luconi, M.; Baldi, E.; Slotte, J.P.; Huhtaniemi, I. Progesterone action in a murine Leydig tumor cell line (mLTC-1), possibly through a nonclassical receptor type. Endocrinology 2000, 141, 247–255. [Google Scholar] [CrossRef]
- Lydon, J.P.; DeMayo, F.J.; Funk, C.R.; Mani, S.K.; Hughes, A.R.; Montgomery, C.A., Jr.; Shyamala, G.; Conneely, O.M.; O’Malley, B.W. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995, 9, 2266–2278. [Google Scholar] [CrossRef]
- Mulac-Jericevic, B.; Lydon, J.P.; DeMayo, F.J.; Conneely, O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci USA 2003, 100, 9744–9749. [Google Scholar] [CrossRef]
- Kananen Rilianawati, K.; Paukku, T.; Markkula, M.; Rainio, E.M.; Huhtaniemi, I. Suppression of gonadotropins inhibits gonadal tumorigenesis in mice transgenic for the mouse inhibin alpha-subunit promoter/simian virus 40 T-antigen fusion gene. Endocrinology 1997, 138, 3521–3531. [Google Scholar] [CrossRef]
- Rahman, N.A.; Kananen Rilianawati, K.; Paukku, T.; Mikola, M.; Markkula, M.; Hamalainen, T.; Huhtaniemi, I.T. Transgenic mouse models for gonadal tumorigenesis. Mol. Cell Endocrinol. 1998, 145, 167–174. [Google Scholar] [CrossRef]
- Kananen, K.; Markkula, M.; el-Hefnawy, T.; Zhang, F.P.; Paukku, T.; Su, J.G.; Hsueh, A.J.; Huhtaniemi, I. The mouse inhibin alpha-subunit promoter directs SV40 T-antigen to Leydig cells in transgenic mice. Mol. Cell Endocrinol. 1996, 119, 135–146. [Google Scholar] [CrossRef]
- Rebois, R.V. Establishment of gonadotropin-responsive murine leydig tumor cell line. J. Cell Biol. 1982, 94, 70–76. [Google Scholar] [CrossRef]
- Heikinheimo, O.; Kontula, K.; Croxatto, H.; Spitz, I.; Luukkainen, T.; Lahteenmaki, P. Plasma concentrations and receptor binding of RU 486 and its metabolites in humans. J. Steroid Biochem. 1987, 26, 279–284. [Google Scholar]
- Sitruk-Ware, R.; Spitz, I.M. Pharmacological properties of mifepristone: Toxicology and safety in animal and human studies. Contraception 2003, 68, 409–420. [Google Scholar] [CrossRef]
- Rice, L.; Waters, C.E.; Eccles, J.; Garside, H.; Sommer, P.; Kay, P.; Blackhall, F.H.; Zeef, L.; Telfer, B.; Stratford, I.; et al. Identification and functional analysis of SKA2 interaction with the glucocorticoid receptor. J. Endocrinol. 2008, 198, 499–509. [Google Scholar] [CrossRef]
- Prefontaine, G.G.; Lemieux, M.E.; Giffin, W.; Schild-Poulter, C.; Pope, L.; LaCasse, E.; Walker, P.; Hache, R.J. Recruitment of octamer transcription factors to DNA by glucocorticoid receptor. Mol. Cell Biol. 1998, 18, 3416–3430. [Google Scholar] [CrossRef]
- Vermeer, H.; Hendriks-Stegeman, B.I.; van der Burg, B.; van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab. 2003, 88, 277–284. [Google Scholar] [CrossRef]
- Chou, J.L.; Chen, L.Y.; Lai, H.C.; Chan, M.W. TGF-beta: Friend or foe? The role of TGF-beta/SMAD signaling in epigenetic silencing of ovarian cancer and its implication in epigenetic therapy. Expert Opin. Ther. Targets 2010, 14, 1213–1223. [Google Scholar] [CrossRef]
- Fauvet, R.; Dufournet Etienne, C.; Poncelet, C.; Bringuier, A.F.; Feldmann, G.; Darai, E. Effects of progesterone and anti-progestin (mifepristone) treatment on proliferation and apoptosis of the human ovarian cancer cell line, OVCAR-3. Oncol. Rep. 2006, 15, 743–748. [Google Scholar] [CrossRef]
- Stringer-Reasor, E.M.; Baker, G.M.; Skor, M.N.; Kocherginsky, M.; Lengyel, E.; Fleming, G.F.; Conzen, S.D. Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2015, 138, 656–662. [Google Scholar] [CrossRef]
- Yu, S.; Yan, C.; Yang, X.; He, S.; Liu, J.; Qin, C.; Huang, C.; Lu, Y.; Tian, Z.; Jia, L. Pharmacoproteomic analysis reveals that metapristone (RU486 metabolite) intervenes E-cadherin and vimentin to realize cancer metastasis chemoprevention. Sci. Rep. 2016, 6, 22388. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Wan, L.; Shao, J.; Lu, Y.; Zhu, Y.; Ou, M.; Yu, S.; Chen, H.; Jia, L. Synthesis, spectral characterization, and in vitro cellular activities of metapristone, a potential cancer metastatic chemopreventive agent derived from mifepristone (RU486). AAPS J. 2014, 16, 289–298. [Google Scholar] [CrossRef]
- Heikinheimo, O.; Lahteenmaki, P.L.; Koivunen, E.; Shoupe, D.; Croxatto, H.; Luukkainen, T.; Lahteenmaki, P. Metabolism and serum binding of RU 486 in women after various single doses. Hum. Reprod. 1987, 2, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Heikinheimo, O.; Ranta, S.; Grunberg, S.; Spitz, I. Alterations in the pituitary-thyroid and pituitary-adrenal axes—Consequences of long-term mifepristone treatment. Metabolism 1997, 46, 292–296. [Google Scholar] [CrossRef]
- Heikinheimo, O.; Haukkamaa, M.; Lahteenmaki, P. Distribution of RU 486 and its demethylated metabolites in humans. J. Clin. Endocrinol. Metab. 1989, 68, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Jewell, C.M.; Bienstock, R.J.; Collins, J.B.; Cidlowski, J.A. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol. Cell Biol. 2007, 27, 2266–2282. [Google Scholar] [CrossRef]
- Kino, T.E.A. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem. Biophys Res. Commun. 2009, 381, 671–675. [Google Scholar] [CrossRef]
- Hales, D.B.; Payne, A.H. Glucocorticoid-mediated repression of P450scc mRNA and de novo synthesis in cultured Leydig cells. Endocrinology 1989, 124, 2099–2104. [Google Scholar] [CrossRef]
- Panza, S.; Malivindi, R.; Chemi, F.; Rago, V.; Giordano, C.; Barone, I.; Bonofiglio, D.; Gelsomino, L.; Giordano, F.; Ando, S.; et al. Glucocorticoid Receptor as a Potential Target to Decrease Aromatase Expression and Inhibit Leydig Tumor Growth. Am. J. Pathol. 2016, 186, 1328–1339. [Google Scholar] [CrossRef]
- Peluso, J.J.; Liu, X.; Saunders, M.M.; Claffey, K.P.; Phoenix, K. Regulation of ovarian cancer cell viability and sensitivity to cisplatin by progesterone receptor membrane component-1. J. Clin. Endocrinol. Metab. 2008, 93, 1592–1599. [Google Scholar] [CrossRef]
- Ahmed, I.S.; Rohe, H.J.; Twist, K.E.; Mattingly, M.N.; Craven, R.J. Progesterone Receptor Membrane Component 1 (Pgrmc1): A Heme-1 Domain Protein That Promotes Tumorigenesis and Is Inhibited by a Small Molecule. J. Pharmacol. Exp. Ther. 2010, 333, 564–573. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.R.; Gonzalez, B.; Rulli, S.B.; Huhtaniemi, I.; Calandra, R.S.; Gonzalez-Calvar, S.I. TGF-beta1 system in Leydig cells. Part I: Effect of hCG and progesterone. J. Reprod. Dev. 2010, 56, 389–395. [Google Scholar] [CrossRef]
- Lebrin, F.; Deckers, M.; Bertolino, P.; Ten Dijke, P. TGF-beta receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Willibald, M.; Bayer, G.; Stahlhut, V.; Poschmann, G.; Stuhler, K.; Gierke, B.; Pawlak, M.; Seeger, H.; Mueck, A.O.; Niederacher, D.; et al. Progesterone receptor membrane component 1 is phosphorylated upon progestin treatment in breast cancer cells. Oncotarget 2017, 8, 72480–72493. [Google Scholar] [CrossRef] [PubMed]
- Roberto, D.; Selvarajah, S.; Park, P.C.; Berman, D.; Venkateswaran, V. Functional validation of metabolic genes that distinguish Gleason 3 from Gleason 4 prostate cancer foci. Prostate 2019, 79, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Peluso, J.J.; Romak, J.; Liu, X. Progesterone receptor membrane component-1 (PGRMC1) is the mediator of progesterone’s antiapoptotic action in spontaneously immortalized granulosa cells as revealed by PGRMC1 small interfering ribonucleic acid treatment and functional analysis of PGRMC1 mutations. Endocrinology 2008, 149, 534–543. [Google Scholar] [CrossRef]
- Ruan, X.; Zhang, Y.; Mueck, A.O.; Willibald, M.; Seeger, H.; Fehm, T.; Brucker, S.; Neubauer, H. Increased expression of progesterone receptor membrane component 1 is associated with aggressive phenotype and poor prognosis in ER-positive and negative breast cancer. Menopause 2017, 24, 203–209. [Google Scholar] [CrossRef]
- Study of Oral Mifepristone as Salvage Therapy in Patients With Advanced or Metastatic Non-Small Cell Lung Cancer. Available online: https://ClinicalTrials.gov/show/NCT02642939 (accessed on 27 July 2020).
- Enzalutamide and Mifepristone in Treating Patients with Metastatic Hormone Resistant Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT02012296 (accessed on 27 July 2020).
- BRCA1/2 and Effect of Mifepristone on the Breast. Available online: https://ClinicalTrials.gov/show/NCT01898312 (accessed on 27 July 2020).
- Haavisto, A.M.; Pettersson, K.; Bergendahl, M.; Perheentupa, A.; Roser, J.F.; Huhtaniemi, I. A supersensitive immunofluorometric assay for rat luteinizing hormone. Endocrinology 1993, 132, 1687–1691. [Google Scholar] [CrossRef]
- Van Casteren, J.I.; Schoonen, W.G.; Kloosterboer, H.J. Development of time-resolved immunofluorometric assays for rat follicle-stimulating hormone and luteinizing hormone and application on sera of cycling rats. Biol. Reprod. 2000, 62, 886–894. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponikwicka-Tyszko, D.; Chrusciel, M.; Pulawska, K.; Bernaczyk, P.; Sztachelska, M.; Guo, P.; Li, X.; Toppari, J.; Huhtaniemi, I.T.; Wołczyński, S.; et al. Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice. Cancers 2020, 12, 3263. https://doi.org/10.3390/cancers12113263
Ponikwicka-Tyszko D, Chrusciel M, Pulawska K, Bernaczyk P, Sztachelska M, Guo P, Li X, Toppari J, Huhtaniemi IT, Wołczyński S, et al. Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice. Cancers. 2020; 12(11):3263. https://doi.org/10.3390/cancers12113263
Chicago/Turabian StylePonikwicka-Tyszko, Donata, Marcin Chrusciel, Kamila Pulawska, Piotr Bernaczyk, Maria Sztachelska, Peilan Guo, Xiangdong Li, Jorma Toppari, Ilpo T. Huhtaniemi, Slawomir Wołczyński, and et al. 2020. "Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice" Cancers 12, no. 11: 3263. https://doi.org/10.3390/cancers12113263
APA StylePonikwicka-Tyszko, D., Chrusciel, M., Pulawska, K., Bernaczyk, P., Sztachelska, M., Guo, P., Li, X., Toppari, J., Huhtaniemi, I. T., Wołczyński, S., & Rahman, N. A. (2020). Mifepristone Treatment Promotes Testicular Leydig Cell Tumor Progression in Transgenic Mice. Cancers, 12(11), 3263. https://doi.org/10.3390/cancers12113263