The Effects of Single Nucleotide Polymorphisms in Cancer RNAi Therapies

 , and
, and 
Simple Summary
Abstract
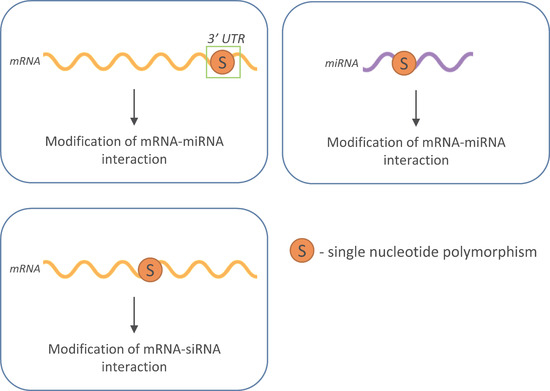
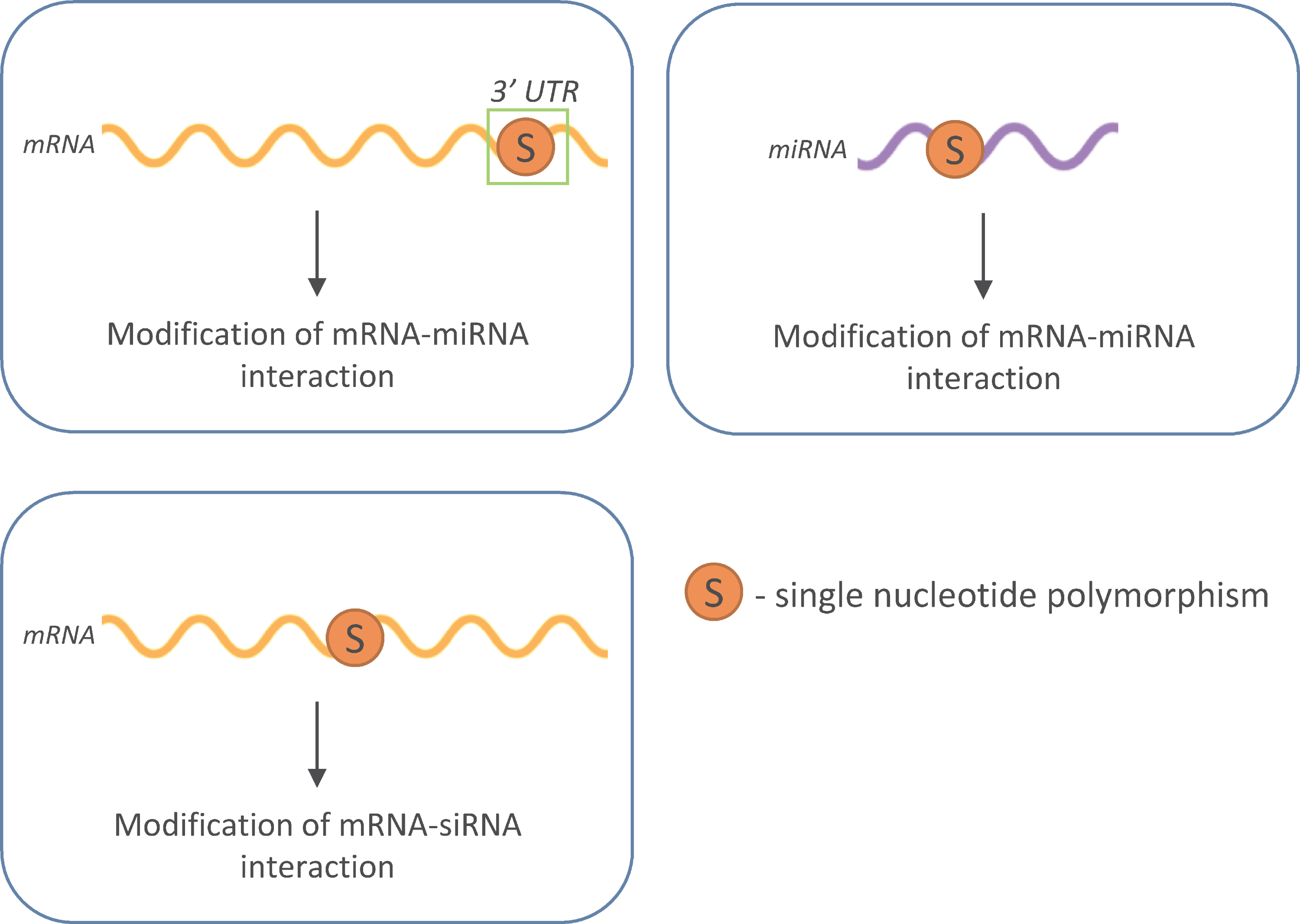
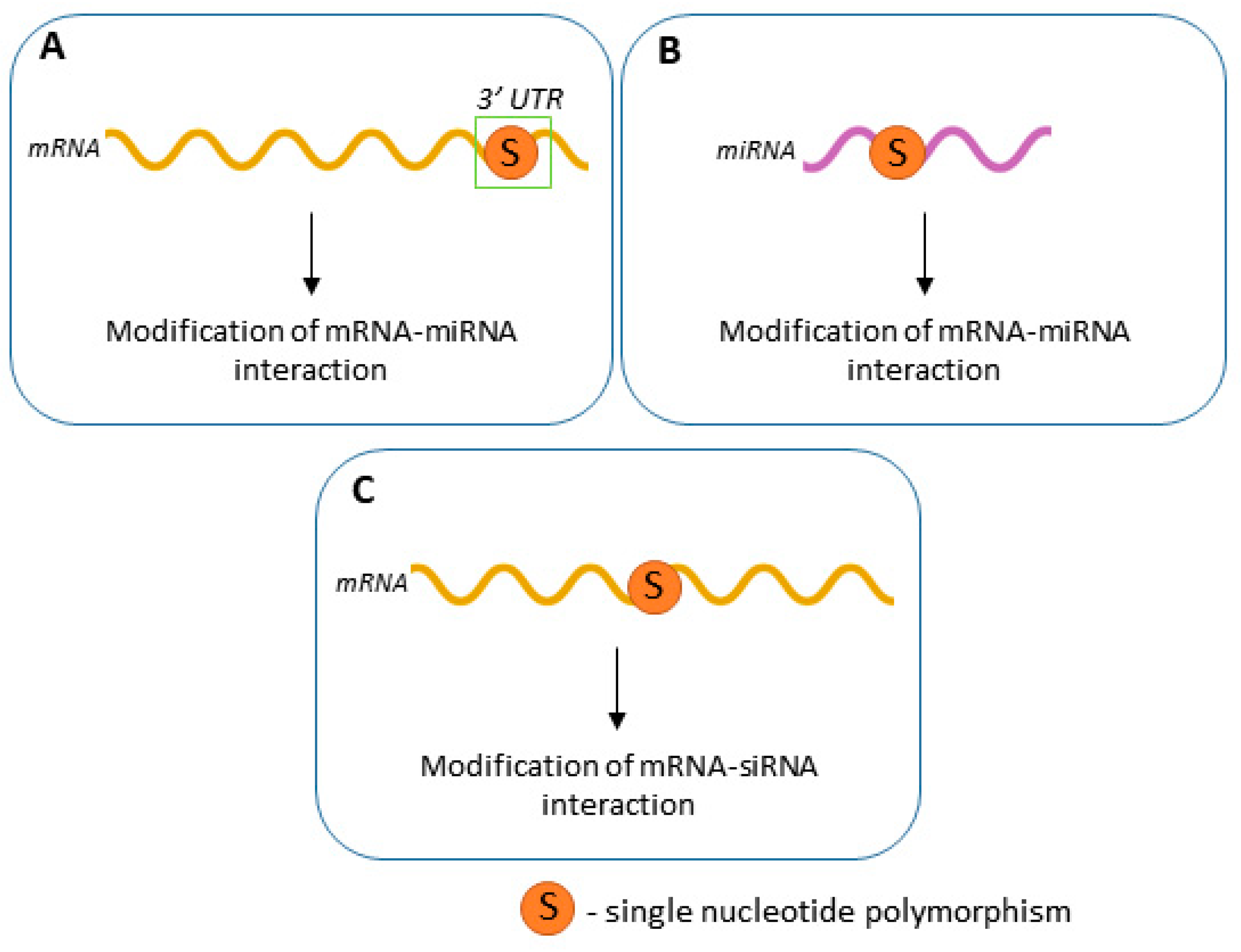
1. Introduction
2. RNA Interference
3. SNPs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Croce, C.M. Molecular origins of cancer: Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Hambly, K.; Danzer, J.; Muskal, S.; Debe, D.A. Interrogating the druggable genome with structural informatics. Mol. Divers. 2006, 10, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar]
- Dimitrov, D.S.; Marks, J.D. Therapeutic antibodies: Current state and future trends—Is a paradigm change coming soon? Methods Mol. Biol. 2009, 525, 1–27. [Google Scholar]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.Q.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Zhong, X.M.; Zhang, D.M.; Xiong, M.M.; Zhang, L. Noncoding RNA for cancer gene therapy. Curr. Strateg. Cancer Gene Ther. 2016, 209, 51–60. [Google Scholar]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; et al. Sequence- and target-independent angiogenesis suppression by siRNA via Tlr3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef]
- DeVincenzo, J.P.; Wilkinson, T.; Vaishnaw, A.; Cehelsky, J.; Meyers, R.; Nochur, S.; Harrison, L.; Meeking, P.; Mann, A.; Moane, E.; et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2010, 182, 1305–1314. [Google Scholar] [CrossRef]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2010, 107, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ackley, K.L. Are we there yet? An update on oligonucleotide drug development. Chim Oggi 2016, 34, Xxxv–Xxxviii. [Google Scholar]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ajroud-Driss, S.; Conceicao, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Scott, L.J. Givosiran: First approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- Wang, X.; Xu, X.; Ma, Z.; Huo, Y.; Xiao, Z.; Li, Y.; Wang, Y. Dynamic mechanisms for pre-miRNA binding and export by exportin-5. RNA 2011, 17, 1511–1528. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi therapies: Drugging the undruggable. Sci. Transl. Med. 2014, 6, 240ps7. [Google Scholar] [CrossRef]
- Fakhr, E.; Zare, F.; Teimoori-Toolabi, L. Precise and efficient siRNA design: A key point in competent gene silencing. Cancer Gene 2016, 23, 73–82. [Google Scholar] [CrossRef]
- Sethupathy, P.; Collins, F.S. Microrna target site polymorphisms and human disease. Trends Genet. TIG 2008, 24, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Moszynska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. Snps in microrna target sites and their potential role in human disease. Open Biol. 2017, 7, 170019. [Google Scholar] [CrossRef]
- Kroliczewski, J.; Sobolewska, A.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. MicroRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene 2018, 640, 66–72. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Kroliczewski, J.; Piotrowski, A.; Jasiecka, A.J.; Bartoszewska, S.; Vecchio-Pagan, B.; Fu, L.W.; Sobolewska, A.; Matalon, S.; Cutting, G.R.; et al. Codon bias and the folding dynamics of the cystic fibrosis transmembrane conductance regulator. Cell. Mol. Biol. Lett. 2016, 21, 23. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, R. Prediction of deleterious nonsynonymous single-nucleotide polymorphism for human diseases. Sci. World J. 2013, 2013, 675851. [Google Scholar] [CrossRef]
- Shastry, B.S. SNPs in disease gene mapping, medicinal drug development and evolution. J. Hum. Genet. 2007, 52, 871–880. [Google Scholar] [CrossRef]
- Fernandez, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from bench to clinic as next generation medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.-W. SiRNA therapeutics: A clinical reality. Sci. China Life Sci. 2020, 63, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Aghamiri, S.; Jafarpour, A.; Malekshahi, Z.V.; Mahmoudi Gomari, M.; Negahdari, B. Targeting siRNA in colorectal cancer therapy: Nanotechnology comes into view. J. Cell. Physiol. 2019, 234, 14818–14827. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Allshire, R.C. RNA silencing and genome regulation. Trends Cell Biol. 2005, 15, 251–258. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Sikorski, A.F. Editorial focus: Entering into the non-coding RNA era. Cell. Mol. Biol. Lett. 2018, 23, 45. [Google Scholar] [CrossRef]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Czech, B.; Hannon, G.J. One loop to rule them all: The ping-pong cycle and piRNA-guided silencing. Trends Biochem. Sci. 2016, 41, 324–337. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Sikorski, A.F. Editorial focus: Understanding off-target effects as the key to successful RNAi therapy. Cell. Mol. Biol. Lett. 2019, 24, 1–23. [Google Scholar] [CrossRef]
- Agrawal, N.; Dasaradhi, P.V.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. MMBR 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Tang, G. Sirna and mirna: An insight into riscs. Trends Biochem. Sci. 2005, 30, 106–114. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. SiRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of MicroRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. Posttranscriptional regulation of microRNA biogenesis in animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef]
- Davis, B.N.; Hata, A. Regulation of microRNA biogenesis: A miRiad of mechanisms. Cell Commun. Signal. CCS 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.W.; Farh, K.K.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Eystathioy, T.; Chan, E.K.; Tenenbaum, S.A.; Keene, J.D.; Griffith, K.; Fritzler, M.J. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol. Biol. Cell 2002, 13, 1338–1351. [Google Scholar] [CrossRef]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef]
- Van Dijk, E.; Cougot, N.; Meyer, S.; Babajko, S.; Wahle, E.; Seraphin, B. Human Dcp2: A catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002, 21, 6915–6924. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. SiRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Rojas-Rios, P.; Simonelig, M. PiRNAs and piwi proteins: Regulators of gene expression in development and stem cells. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Rouget, C.; Papin, C.; Boureux, A.; Meunier, A.C.; Franco, B.; Robine, N.; Lai, E.C.; Pelisson, A.; Simonelig, M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early drosophila embryo. Nature 2010, 467, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Barckmann, B.; Pierson, S.; Dufourt, J.; Papin, C.; Armenise, C.; Port, F.; Grentzinger, T.; Chambeyron, S.; Baronian, G.; Desvignes, J.P.; et al. Aubergine iclip reveals piRNA-dependent decay of mRNAs involved in germ cell development in the early embryo. Cell Rep. 2015, 12, 1205–1216. [Google Scholar] [CrossRef]
- Sivagurunathan, S.; Arunachalam, J.P.; Chidambaram, S. PIWI-like protein, HIWI2 is aberrantly expressed in retinoblastoma cells and affects cell-cycle potentially through OTX2. Cell. Mol. Biol. Lett. 2017, 22, 17. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Yan, K.W.; Liu, C.Y.; Li, P.F.; Wang, K. PIWI family emerging as a decisive factor of cell fate: An overview. Eur. J. Cell Biol. 2017, 96, 746–757. [Google Scholar] [CrossRef]
- Burroughs, A.M.; Iyer, L.M.; Aravind, L. Two novel piwi families: Roles in inter-genomic conflicts in bacteria and mediator-dependent modulation of transcription in eukaryotes. Biol. Direct 2013, 8, 13. [Google Scholar] [CrossRef]
- Kwon, C.; Tak, H.; Rho, M.; Chang, H.R.; Kim, Y.H.; Kim, K.T.; Balch, C.; Lee, E.K.; Nam, S. Detection of PIWI and piRNAs in the mitochondria of mammalian cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 218–223. [Google Scholar] [CrossRef]
- Suzuki, R.; Honda, S.; Kirino, Y. PIWI expression and function in cancer. Front. Genet. 2012, 3, 204. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Hubaux, R.; Enfield, K.S.; Pikor, L.A.; Becker-Santos, D.D.; Brown, C.J.; Lam, S.; Lam, W.L. Unique somatic and malignant expression patterns implicate PIWI-interacting RNAs in cancer-type specific biology. Sci. Rep. 2015, 5, 10423. [Google Scholar] [CrossRef]
- Mei, Y.; Wang, Y.; Kumari, P.; Shetty, A.C.; Clark, D.; Gable, T.; MacKerell, A.D.; Ma, M.Z.; Weber, D.J.; Yang, A.J.; et al. A piRNA-like small RNA interacts with and modulates p-ERM proteins in human somatic cells. Nat. Commun. 2015, 6, 7316. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Clark, D.; Mao, L. Novel dimensions of piRNAs in cancer. Cancer Lett. 2013, 336, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gebert, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Moszynska, A.; Cabaj, A.; Kroliczewski, J.; Madanecki, P.; Ochocka, R.J.; Crossman, D.K.; Collawn, J.F.; et al. PIWI proteins contribute to apoptosis during the UPR in human airway epithelial cells. Sci. Rep. 2018, 8, 16431. [Google Scholar] [CrossRef]
- Saberi, F.; Kamali, M.; Najafi, A.; Yazdanparast, A.; Moghaddam, M.M. Natural antisense RNAs as mRNA regulatory elements in bacteria: A review on function and applications. Cell. Mol. Biol. Lett. 2016, 21, 6. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Ishizu, H.; Siomi, H.; Siomi, M.C. Biology of PIWI-interacting RNAs: New insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012, 26, 2361–2373. [Google Scholar] [CrossRef]
- Erhard, F.; Zimmer, R. Classification of ncRNAs using position and size information in deep sequencing data. Bioinformatics 2010, 26, i426–i432. [Google Scholar] [CrossRef]
- Szell, M.; Bata-Csorgo, Z.; Kemeny, L. The enigmatic world of mRNA-like ncRNAs: Their role in human evolution and in human diseases. Semin. Cancer Biol. 2008, 18, 141–148. [Google Scholar] [CrossRef]
- Huang, R.; Jaritz, M.; Guenzl, P.; Vlatkovic, I.; Sommer, A.; Tamir, I.M.; Marks, H.; Klampfl, T.; Kralovics, R.; Stunnenberg, H.G.; et al. An RNA-seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS ONE 2011, 6, e27288. [Google Scholar] [CrossRef]
- Pereira, D.M.; Rodrigues, P.M.; Borralho, P.M.; Rodrigues, C.M. Delivering the promise of miRNA cancer therapeutics. Drug Discov. Today 2013, 18, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Liu, D.; Lai, H.; Li, J.; Wang, C. Developing miRNA therapeutics for cardiac repair in ischemic heart disease. J. Thorac. Dis. 2016, 8, E918–E927. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Laursen, M.B.; Nielsen, A.F.; Hansen, T.B.; Bus, C.; Langkjaer, N.; Babu, B.R.; Hojland, T.; Abramov, M.; Van Aerschot, A.; et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009, 37, 2867–2881. [Google Scholar] [CrossRef] [PubMed]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release Off. J. Control. Release Soc. 2014, 195, 49–54. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Ehlert, E.M.; Eggers, R.; Niclou, S.P.; Verhaagen, J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 2010, 11, 20. [Google Scholar] [CrossRef]
- Davidson, B.L.; McCray, P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Carlin, C.R. New insights to adenovirus-directed innate immunity in respiratory epithelial cells. Microorganisms 2019, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate immunity to adenovirus. Hum. Gene 2014, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; Liu, J.; Li, H.; Walsh, S.; Johnson, J.; Milner, D.; Seaman, M.; Krause, K.; Swan, E.; Tucker, R.; et al. A phase1 clinical trial to evaluate the safety, mucosal and innate immunity of adenovirus type 26 HIV-1 vaccine in healthy, HIV-1 uninfected adults. Aids Res. Hum. Retrov. 2011, 27, A124. [Google Scholar]
- Baum, C.; Kustikova, O.; Modlich, U.; Li, Z.; Fehse, B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum. Gene 2006, 17, 253–263. [Google Scholar]
- Pauwels, K.; Gijsbers, R.; Toelen, J.; Schambach, A.; Willard-Gallo, K.; Verheust, C.; Debyser, Z.; Herman, P. State-of-the-art lentiviral vectors for research use: Risk assessment and biosafety recommendations. Curr. Gene 2009, 9, 459–474. [Google Scholar] [CrossRef]
- Seow, Y.; Wood, M.J. Biological gene delivery vehicles: Beyond viral vectors. Mol. Ther. 2009, 17, 767–777. [Google Scholar] [CrossRef]
- Polansky, H.; Schwab, H. Latent viruses can cause disease by disrupting the competition for the limiting factor p300/cbp. Cell. Mol. Biol. Lett. 2018, 23, 56. [Google Scholar] [CrossRef]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
- Kwekkeboom, R.F.; Lei, Z.; Doevendans, P.A.; Musters, R.J.; Sluijter, J.P. Targeted delivery of miRNA therapeutics for cardiovascular diseases: Opportunities and challenges. Clin. Sci. 2014, 127, 351–365. [Google Scholar] [CrossRef]
- Petri, S.; Meister, G. SiRNA design principles and off-target effects. Methods Mol. Biol. 2013, 986, 59–71. [Google Scholar] [PubMed]
- Wyrozumska, P.; Meissner, J.; Toporkiewicz, M.; Szarawarska, M.; Kuliczkowski, K.; Ugorski, M.; Walasek, M.A.; Sikorski, A.F. Liposome-coated lipoplex-based carrier for antisense oligonucleotides. Cancer Biol. Ther. 2015, 16, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.M.; Toporkiewicz, M.; Czogalla, A.; Matusewicz, L.; Kuliczkowski, K.; Sikorski, A.F. Novel antisense therapeutics delivery systems: In vitro and in vivo studies of liposomes targeted with anti-cd20 antibody. J. Control. Release 2015, 220, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Wyrozumska, P.; Stebelska, K.; Grzybek, M.; Sikorski, A.F. Synthetic vectors for genetic drug delivery. In Nanocarrier Technologies: Frontiers of Nanotherapy; Mozafari, M.R., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 139–174. [Google Scholar]
- Tseng, Y.C.; Mozumdar, S.; Huang, L. Lipid-based systemic delivery of siRNA. Adv. Drug Deliv. Rev. 2009, 61, 721–731. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Neuberg, P.; Kichler, A. Recent developments in nucleic acid delivery with polyethylenimines. Adv. Genet. 2014, 88, 263–288. [Google Scholar]
- Urban-Klein, B.; Werth, S.; Abuharbeid, S.; Czubayko, F.; Aigner, A. Rnai-mediated gene-targeting through systemic application of polyethylenimine (pei)-complexed siRNA in vivo. Gene 2005, 12, 461–466. [Google Scholar] [CrossRef]
- Somani, S.; Laskar, P.; Altwaijry, N.; Kewcharoenvong, P.; Irving, C.; Robb, G.; Pickard, B.S.; Dufes, C. Pegylation of polypropylenimine dendrimers: Effects on cytotoxicity, DNA condensation, gene delivery and expression in cancer cells. Sci. Rep. 2018, 8, 9410. [Google Scholar] [CrossRef]
- Peng, L. Dendrimers as nanovectors for RNA delivery in gene therapy. Eur. J. Pharm. Sci. 2013, 50, E31. [Google Scholar]
- Dufes, C.; Uchegbu, I.F.; Schatzlein, A.G. Dendrimers in gene delivery. Adv. Drug Deliv. Rev. 2005, 57, 2177–2202. [Google Scholar] [CrossRef] [PubMed]
- Shcharbin, D.; Shakhbazau, A.; Bryszewska, M. Poly(amidoamine) dendrimer complexes as a platform for gene delivery. Expert Opin. Drug Deliv. 2013, 10, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, G.; Su, Z.; Jiang, Z.; Chen, L.; Wang, J.; Yu, S.; Liu, Z. Poly(amido amine) is an ideal carrier of miR-7 for enhancing gene silencing effects on the EGFR pathway in u251 glioma cells. Oncol. Rep. 2013, 29, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Taratula, O.R.; Rodriguez-Rodriguez, L.; Minko, T. Targeted nanomedicine for suppression of cd44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of siRNA and anticancer drug. Clin. Cancer Res. 2013, 19, 6193–6204. [Google Scholar] [CrossRef]
- Siu, K.S.; Chen, D.; Zheng, X.; Zhang, X.; Johnston, N.; Liu, Y.; Yuan, K.; Koropatnick, J.; Gillies, E.R.; Min, W.P. Non-covalently functionalized single-walled carbon nanotube for topical siRNA delivery into melanoma. Biomaterials 2014, 35, 3435–3442. [Google Scholar] [CrossRef]
- Chien, C.-S.; Chiou, S.-H. Abstract 3321: Cationic polyurethanes-short branch pei-mediated delivery of miR145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. Cancer Res. 2012, 72, 3321. [Google Scholar]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Bartlett, D.W.; Davis, M.E. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol. Bioeng. 2008, 99, 975–985. [Google Scholar] [CrossRef]
- Videira, M.; Arranja, A.; Rafael, D.; Gaspar, R. Preclinical development of siRNA therapeutics: Towards the match between fundamental science and engineered systems. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 689–702. [Google Scholar] [CrossRef]
- Bitar, A.; Ahmad, N.M.; Fessi, H.; Elaissari, A. Silica-based nanoparticles for biomedical applications. Drug Discov. Today 2012, 17, 1147–1154. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Liu, T.; Zhang, D.S.; Wang, Y.; Gu, H.; Di, W. Highly effective antiangiogenesis via magnetic mesoporous silica-based siRNA vehicle targeting the VEGF gene for orthotopic ovarian cancer therapy. Int. J. Nanomed. 2015, 10, 2579–2594. [Google Scholar]
- Chen, Y.; Gu, H.; Zhang, D.S.; Li, F.; Liu, T.; Xia, W. Highly effective inhibition of lung cancer growth and metastasis by systemic delivery of siRNA via multimodal mesoporous silica-based nanocarrier. Biomaterials 2014, 35, 10058–10069. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Sevilla, I.; Artiga, A.; Mitchell, S.G.; De Matteis, L.; de la Fuente, J.M. Natural polysaccharides for siRNA delivery: Nanocarriers based on chitosan, hyaluronic acid, and their derivatives. Molecules 2019, 24, 2570. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.; Hobel, S.; Bakowsky, U.; Aigner, A. Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials 2010, 31, 6892–6900. [Google Scholar] [CrossRef]
- Jang, Y.L.; Yun, U.J.; Lee, M.S.; Kim, M.G.; Son, S.; Lee, K.; Chae, S.Y.; Lim, D.W.; Kim, H.T.; Kim, S.H.; et al. Cell-penetrating peptide mimicking polymer-based combined delivery of paclitaxel and siRNA for enhanced tumor growth suppression. Int. J. Pharm. 2012, 434, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Alshamsan, A.; Hamdy, S.; Samuel, J.; El-Kadi, A.O.; Lavasanifar, A.; Uludag, H. The induction of tumor apoptosis in b16 melanoma following stat3 siRNA delivery with a lipid-substituted polyethylenimine. Biomaterials 2010, 31, 1420–1428. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapy silencing an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Zhou, J.H.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of thiomab-siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Kim, N.Y.; Choi, Y.B.; Park, S.H.; Yang, J.M.; Shin, S. Rna interference in vitro and in vivo using an arginine peptide/sirna complex system. J. Control. Release 2010, 143, 335–343. [Google Scholar] [CrossRef]
- Saxena, S.; Jonsson, Z.O.; Dutta, A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 2003, 278, 44312–44319. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Scacheri, P.C.; Rozenblatt-Rosen, O.; Caplen, N.J.; Wolfsberg, T.G.; Umayam, L.; Lee, J.C.; Hughes, C.M.; Shanmugam, K.S.; Bhattacharjee, A.; Meyerson, M.; et al. Short interfering RNAs can induce unexpected and divergent changes in the levels of untargeted proteins in mammalian cells. Proc Natl. Acad. Sci. USA 2004, 101, 1892–1897. [Google Scholar] [CrossRef]
- Erichsen, H.C.; Chanock, S.J. Snps in cancer research and treatment. Br. J. Cancer 2004, 90, 747–751. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Jiang, M.; Tian, W.; Zhou, B. Single nucleotide polymorphisms in PLCE1 for cancer risk of different types: A meta-analysis. Front. Oncol. 2018, 8, 613. [Google Scholar] [CrossRef]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef]
- Yang, J.-P.; Wang, W.-B.; Yang, X.-X.; Yang, L.; Ren, L.; Zhou, F.-X.; Hu, L.; He, W.; Li, B.-Y.; Zhu, Y.; et al. The mpo−463g>a polymorphism and lung cancer risk: A meta-analysis based on 22 case–control studies. PLoS ONE 2013, 8, e65778. [Google Scholar] [CrossRef]
- Li, J.; Fu, Y.; Zhao, B.; Xiao, Y.; Chen, R. Myeloperoxidase g463a polymorphism and risk of lung cancer. Tumor Biol. 2014, 35, 821–829. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Gisselsson, D.; Dumanski, J.P. Mosaicism in health and disease—Clones picking up speed. Nat. Rev. Genet. 2017, 18, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Fattah, A.A.; Hamid Sadik, N.A.; Shaker, O.G.; Mohamed Kamal, A. Single nucleotide polymorphism in smad7 and chi3l1 and colorectal cancer risk. In Mediators of Inflammation; Hindawi Limited: London, UK, 2018; Volume 2018. [Google Scholar]
- Collins, F.S.; Brooks, L.D.; Chakravarti, A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998, 8, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Shatoff, E.; Bundschuh, R. Single nucleotide polymorphisms affect RNA-protein interactions at a distance through modulation of RNA secondary structures. PLoS Comput. Biol. 2020, 16, e1007852. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. Microrna regulation by rna-binding proteins and its implications for cancer. In Nature Reviews Cancer; Nature Publishing Group: Berlin, Germany, 2011; Volume 11, pp. 644–656. [Google Scholar]
- Mansfield, K.D.; Keene, J.D. The ribonome: A dominant force in co-ordinating gene expression. Biol. Cell 2009, 101, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 1994, 265, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.A.; Jablonsky, M.; Bartoszewska, S.; Stevenson, L.; Dai, Q.; Kappes, J.; Collawn, J.F.; Bebok, Z. A synonymous single nucleotide polymorphism in delta f508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biol. Chem. 2010, 285, 28741–28748. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Taioli, E.; Benhamou, S.; Bouchardy, C.; Cascorbi, I.; Cajas-Salazar, N.; Dally, H.; Fong, K.M.; Larsen, J.E.; Le Marchand, L.; London, S.J.; et al. Myeloperoxidase g-463a polymorphism and lung cancer: A huge genetic susceptibility to environmental carcinogens pooled analysis. Genet. Med. 2007, 9, 67–73. [Google Scholar] [CrossRef]
- Hsu, P.-I.; Jwo, J.-J.; Yang, C.-L.; Hsu, P.-N.; Yang, H.-B.; Lai, K.-H.; Chen, I.-S.; Chuah, S.-K.; Wu, D.-C.; Chen, A. Association of the myeloperoxidase polymorphism with the risk of gastric cancer. Anticancer Res. 2008, 28, 1317–1323. [Google Scholar] [PubMed]
- Tafer, H. Bioinformatics of siRNA Design; Humana Press: Totowa, NJ, USA, 2014; pp. 477–490. [Google Scholar]
- Peltoketo, H.; Piao, Y.; Mannermaa, A.; Ponder, B.A.J.; Isomaa, V.; Poutanen, M.; Winqvist, R.; Vihko, R. A point mutation in the putative tata box, detected in nondiseased individuals and patients with hereditary breast cancer, decreases promoter activity of the 17β-hydroxysteroid dehydrogenase type 1 gene 2 (edh17b2) in vitro. Genomics 1994, 23, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.A.; Kondo, Y.; Chen, X.; Shen, L.; Guo, Y.; Tellez, C.; Estécio, M.R.H.; Ahmed, S.; Issa, J.-P.J. An sp1/sp3 binding polymorphism confers methylation protection. PLoS Genet. 2008, 4, e1000162. [Google Scholar] [CrossRef]
- Butcher, D.T.; Mancini-DiNardo, D.N.; Archer, T.K.; Rodenhiser, D.I. DNA binding sites for putative methylation boundaries in the unmethylated region of thebrca1 promoter. Int. J. Cancer 2004, 111, 669–678. [Google Scholar] [CrossRef]
- Shan, Y.; Eastwood, M.P.; Zhang, X.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef]
- Yates, C.M.; Sternberg, M.J.E. The effects of non-synonymous single nucleotide polymorphisms (nssnps) on protein-protein interactions. J. Mol. Biol. 2013, 425, 3949–3963. [Google Scholar] [CrossRef]
- Scodes, S.; Cappuzzo, F. Determining the appropriate treatment for different EGFR mutations in non-small cell lung cancer patients. Expert Rev. Respir. Med. 2020, 14, 565–576. [Google Scholar] [CrossRef]
- Raghav, D.; Sharma, V.; Agarwal, S.M. Structural investigation of deleterious non-synonymous SNPS of EGFR gene. Interdiscip. Sci. Comput. Life Sci. 2013, 5, 60–68. [Google Scholar] [CrossRef]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (synonymous) SNPS: Should we care about them? Method Mol. Biol. 2009, 78, 23–39. [Google Scholar]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C.; et al. Kras gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Cepero, V.; Sierra, J.R.; Corso, S.; Ghiso, E.; Casorzo, L.; Perera, T.; Comoglio, P.M.; Giordano, S. Met and KRAS gene amplification mediates acquired resistance to Met tyrosine kinase inhibitors. Cancer Res. 2010, 70, 7580–7590. [Google Scholar] [CrossRef]
- Birkeland, E.; Wik, E.; Mjos, S.; Hoivik, E.A.; Trovik, J.; Werner, H.M.J.; Kusonmano, K.; Petersen, K.; Raeder, M.B.; Holst, F.; et al. KRAS gene amplification and overexpression but not mutation associates with aggressive and metastatic endometrial cancer. Br. J. Cancer 2012, 107, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Groß, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Mita, H.; Toyota, M.; Aoki, F.; Akashi, H.; Maruyama, R.; Sasaki, Y.; Suzuki, H.; Idogawa, M.; Kashima, L.; Yanagihara, K.; et al. A novel method, digital genome scanning detects KRAS gene amplification in gastric cancers: Involvement of overexpressed wild-type KRAS in downstream signaling and cancer cell growth. BMC Cancer 2009, 9, 1–16. [Google Scholar] [CrossRef]
- Liu, W.; Han, B.; Zhu, W.; Cheng, T.; Fan, M.; Wu, J.; Yang, Y.; Zhu, H.; Si, J.; Lyu, Q.; et al. Polymorphism in the alternative donor site of the cryptic exon of LHCGR: Functional consequences and associations with testosterone level. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Broen, K.; Levenga, H.; Vos, J.; van Bergen, K.; Fredrix, H.; Greupink-Draaisma, A.; Kester, M.; Falkenburg, J.H.F.; de Mulder, P.; de Witte, T.; et al. A polymorphism in the splice donor site of znf419 results in the novel renal cell carcinoma-associated minor histocompatibility antigen zaphir. PLoS ONE 2011, 6, e21699. [Google Scholar] [CrossRef] [PubMed]
- Kurmangaliyev, Y.Z.; Sutormin, R.A.; Naumenko, S.A.; Bazykin, G.A.; Gelfand, M.S. Functional implications of splicing polymorphisms in the human genome. Hum. Mol. Genet. 2013, 22, 3449–3459. [Google Scholar] [CrossRef]
- Bonnevie-Nielsen, V.; Field, L.L.; Lu, S.; Zheng, D.J.; Li, M.; Martensen, P.M.; Nielsen, T.B.; Beck-Nielsen, H.; Lau, Y.L.; Pociot, F. Variation in antiviral 2′,5′-oligoadenylate synthetase (2′5′as) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the oas1 gene. Am. J. Hum. Genet. 2005, 76, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, E.; Ha, K.C.H.; Wang, Z.B.; Bemmo, A.; Kleinman, C.L.; Kwan, T.; Pastinen, T.; Majewski, J. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 2011, 21, 545–554. [Google Scholar] [CrossRef]
- Gong, J.; Liu, W.; Zhang, J.; Miao, X.; Guo, A.Y. lncRNASNP: A database of SNPS in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. 2015, 43, D181–D186. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.H.; Lu, Q.; Ouyang, Y.D.; Mao, H.L.; Zhang, P.B.; Yao, J.L.; Xu, C.G.; Li, X.H.; Xiao, J.H.; Zhang, Q.F. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef]
- Wang, R.; Feng, N.; Wang, Y.; Gao, S.; Zhang, F.; Qian, Y.; Gao, M.; Yu, H.; Zhou, B.; Qian, B. SNPs in lncRNA genes are associated with non-small cell lung cancer in a Chinese population. J. Clin. Lab. Anal. 2019, 33, e22858. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Feng, N.; Zhu, T.; Li, Q.; Zhang, Q.; Wang, Y.; Gao, M.; Zhou, B.; Yu, H.; Zheng, M.; et al. A SNP-mediated lncRNA (loc146880) and microRNA (mir-539-5p) interaction and its potential impact on the NSCLC risk. J. Exp. Clin. Cancer Res. 2020, 39, 157. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA hotair reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding rna malat1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Shang, C.; Guo, Y.; Zhang, H.; Xue, Y.X. Long noncoding rna hotair is a prognostic biomarker and inhibits chemosensitivity to doxorubicin in bladder transitional cell carcinoma. Cancer Chemother. Pharmacol. 2016, 77, 507–513. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Ding, H.; Kennington, L.; Moore, J.T.; Schelter, J.; Burchard, J.; Linsley, P.S.; Aronin, N.; Xu, Z.; Zamore, P.D. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006, 2, e140. [Google Scholar] [CrossRef]
- Ding, H.; Schwarz, D.S.; Keene, A.; Affar, E.B.; Fenton, L.; Xia, X.; Shi, Y.; Zamore, P.D.; Xu, Z. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003, 2, 209–217. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Gouvion, C.M.; Davidson, B.L.; Paulson, H.L. Targeting Alzheimer’s disease genes with RNA interference: An efficient strategy for silencing mutant alleles. Nucleic Acids Res. 2004, 32, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Abdelgany, A.; Wood, M.; Beeson, D. Allele-specific silencing of a pathogenic mutant acetylcholine receptor subunit by RNA interference. Hum. Mol. Genet. 2003, 12, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Xia, H.; Marrs, G.L.; Gouvion, C.M.; Lee, G.; Davidson, B.L.; Paulson, H.L. Allele-specific silencing of dominant disease genes. Proc. Natl. Acad. Sci. USA 2003, 100, 7195–7200. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M.; Ang, C.; Dimaio, C.J.; Javle, M.M.; Gutierrez, M.; Yarom, N.; Stemmer, S.M.; Golan, T.; Geva, R.; Semenisty, V.; et al. A phase ii study of sig12d-loder in combination with chemotherapy in patients with locally advanced pancreatic cancer (protact). J. Clin. Oncol. 2020, 38, TPS4672. [Google Scholar] [CrossRef]
- Pantsar, T.; Rissanen, S.; Dauch, D.; Laitinen, T.; Vattulainen, I.; Poso, A. Assessment of mutation probabilities of KRAS g12 missense mutants and their long-timescale dynamics by atomistic molecular simulations and markov state modeling. PLoS Comput. Biol. 2018, 14, e1006458. [Google Scholar] [CrossRef]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. P53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Huang, C.-H.; Wu, W.-J.; Chang, L.-C.; Lung, F.-W. Tp53 codon 72 gene polymorphism paradox in associated with various carcinoma incidences, invasiveness and chemotherapy responses. Int. J. Biomed. Sci. 2008, 4, 248–254. [Google Scholar]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef]
- Frank, A.K.; Leu, J.I.-J.; Zhou, Y.; Devarajan, K.; Nedelko, T.; Klein-Szanto, A.; Hollstein, M.; Murphy, M.E. The codon 72 polymorphism of p53 regulates interaction with NF-κB and transactivation of genes involved in immunity and inflammation. Mol. Cell. Biol. 2011, 31, 1201–1213. [Google Scholar] [CrossRef]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer therapeutic targeting using mutant-p53-specific siRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Harborth, J.; Elbashir, S.M.; Bechert, K.; Tuschl, T.; Weber, K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001, 114, 4557–4565. [Google Scholar]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncol. Basel 2005, 69, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. The role of the VEGF signaling pathway in tumor angiogenesis. In Tumor Angiogenesis a Key Target for Cancer Therapy; Springer: Cham, Switzelrand, 2019; pp. 211–226. [Google Scholar]
- Pardali, E.; Godfrey, R.; Waltenberger, J. Vegf signaling in normal and tumor angiogenesis. Tumor Angiogenesis Regul. 2013, 1–36. [Google Scholar]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef]
- Naikoo, N.A.; Afroze, D.; Rasool, R.; Shah, S.; Ahangar, A.G.; Bhat, I.A.; Qasim, I.; Siddiqi, M.A.; Shah, Z.A. Snp and haplotype analysis of vascular endothelial growth factor (VEGF) gene in lung cancer patients of kashmir. Asian Pac. J. Cancer Prev. 2017, 18, 1799–1804. [Google Scholar]
- Jain, L.; Vargo, C.A.; Danesi, R.; Sissung, T.M.; Price, D.K.; Venzon, D.; Venitz, J.; Figg, W.D. The role of vascular endothelial growth factor SNPs as predictive and prognostic markers for major solid tumors. Mol. Cancer 2009, 8, 2496–2508. [Google Scholar] [CrossRef]
- Kim, Y.J.; Chung, W.C.; Jun, K.H.; Chin, H.M. Genetic polymorphisms of vascular endothelial growth factor (VEGF) associated with gastric cancer recurrence after curative resection with adjuvant chemotherapy. BMC Cancer 2019, 19, 483. [Google Scholar] [CrossRef]
- Yang, F.M.; Qin, Z.Q.; Shao, C.C.; Liu, W.T.; Ma, L.; Shu, Y.Q.; Shen, H. Association between VEGF gene polymorphisms and the susceptibility to lung cancer: An updated meta-analysis. Biomed. Res. Int. 2018, 2018. [Google Scholar] [CrossRef]
- Ratnasari, N.; Nurdjanah, S.; Sadewa, A.H.; Hakimi, M.; Yano, Y. Difference of polymorphism VEGF-gene rs699947 in indonesian chronic liver disease population. PLoS ONE 2017, 12, e0183503. [Google Scholar] [CrossRef]
- Yvamoto, E.Y.; Ferreira, R.F.; Nogueira, V.; Pinhe, M.A.S.; Tenani, G.D.; Andrade, J.G.S.C.; Baitello, M.E.L.; Gregório, M.L.; Fucuta, P.S.; Silva, R.F.; et al. Influence of vascular endothelial growth factor and alpha-fetoprotein on hepatocellular carcinoma. Genet. Mol. Res. GMR 2015, 14, 17453–17462. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Lightfoot, H.L.; Habib, N.A.; Rossi, J.J. Development of mtl-cebpa: Small activating RNA drug for hepatocellular carcinoma. Curr. Pharm. Biotechnol. 2018, 19, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Bristow, M.R.; Port, J.D. Micrornas in heart failure, cardiac transplantation, and myocardial recovery: Biomarkers with therapeutic potential. Curr. Heart Fail. Rep. 2017, 14, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Mellis, D.; Caporali, A. Microrna-based therapeutics in cardiovascular disease: Screening and delivery to the target. Biochem. Soc. Trans. 2018, 46, 11–21. [Google Scholar] [CrossRef]
- Snowhite, I.V.; Allende, G.; Sosenko, J.; Pastori, R.L.; Messinger Cayetano, S.; Pugliese, A. Association of serum micrornas with islet autoimmunity, disease progression and metabolic impairment in relatives at risk of type 1 diabetes. Diabetologia 2017, 60, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Yu, Z.-H.; Zhao, P.; Zhuang, G.-Q.; Wu, Z.-X.; Dang, L.; Li, H.-Z.; Ma, S.-M.; Cui, Z.-Z.; Zhang, G.-P.; et al. Putative roles as oncogene or tumour suppressor of the mid-clustered micrornas in gallid alphaherpesvirus 2 (gahv2) induced marek’s disease lymphomagenesis. J. Gen. Virol. 2017, 98, 1097–1112. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Gurtan, A.M.; Sharp, P.A. The role of miRNAs in regulating gene expression networks. J. Mol. Biol. 2013, 425, 3582–3600. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, Y.; Xie, S.J.; Xu, S.J.; Zhou, H.; Qu, L.H. Argonaute hits-clip decodes microRNA-mRNA interaction maps during heart development. Cardiology 2013, 126, 62. [Google Scholar]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute hits-clip decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef]
- Liang, X.H.; Hart, C.E.; Crooke, S.T. Transfection of sirnas can alter miRNA levels and trigger non-specific protein degradation in mammalian cells. BBA-Gene Regul. Mech. 2013, 1829, 455–468. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Cabaj, A.; Dabrowski, M.; Collawn, J.F.; Bartoszewski, R. Mir-34c-5p modulates x-box-binding protein 1 (xbp1) expression during the adaptive phase of the unfolded protein response. FASEB J. 2019, 33, 11541–11554. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of mrx34, a liposomal mir-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Titze-de-Almeida, R.; David, C.; Titze-de-Almeida, S.S. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm. Res. 2017, 34, 1339–1363. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Thakral, S.; Ghoshal, K. Mir-122 is a unique molecule with great potential in diagnosis, prognosis of liver disease, and therapy both as miRNA mimic and antimir. Curr. Gene Ther. 2015, 15, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Bei, C.; Liu, S.; Yu, X.; Qiu, M.; Tang, B.; Liao, W.; He, S.; Yu, H. Single nucleotide polymorphisms in mir-122 are associated with the risk of hepatocellular carcinoma in a southern Chinese population. Biomed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.E.; Zheng, T.; Yi, C.; Leaderer, D.; Weidhaas, J.; Slack, F.; Zhang, Y.; Paranjape, T.; Zhu, Y. MicroRNA mir-196a-2 and breast cancer: A genetic and epigenetic association study and functional analysis. Cancer Res. 2009, 69, 5970–5977. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liang, J.; Wang, Z.; Tian, T.; Zhou, X.; Chen, J.; Miao, R.; Wang, Y.; Wang, X.; Shen, H. Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Hum. Mutat. 2009, 30, 79–84. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, J.; Tian, T.; Zhou, X.; Gu, H.; Xu, L.; Zeng, Y.; Miao, R.; Jin, G.; Ma, H.; et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J. Clin. Investig. 2008, 118, 2600–2608. [Google Scholar] [CrossRef]
- Hong, M.J.; Choi, Y.Y.; Jang, J.-A.; Jung, H.-J.; Lee, S.Y.; Lee, W.K.; Yoo, S.S.; Lee, J.; Cha, S.I.; Kim, C.H.; et al. Association between genetic variants in pre-microRNAs and survival of early-stage NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2013, 8, 703–710. [Google Scholar] [CrossRef]
- Behlke, M.A. Progress towards in vivo use of siRNAs. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 644–670. [Google Scholar] [CrossRef]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef]
- Stevens, J.L. Future of toxicology—Mechanisms of toxicity and drug safety: Where do we go from here? Chem. Res. Toxicol. 2006, 19, 1393–1401. [Google Scholar] [CrossRef]
- Pentin, J.; Smith, J. Drug calculations: Are they safer with or without a calculator? Br. J. Nurs. 2006, 15, 778–781. [Google Scholar] [CrossRef]
- Smith, D.A.; Schmid, E.F. Drug withdrawals and the lessons within. Curr. Opin. Drug Discov. Dev. 2006, 9, 38–46. [Google Scholar]
- Visconti, J.A.; Smith, M.C. The economics of adverse drug reactions—Case studies. Ann. Pharmacother. 2006, 40, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Brinster, R.; Scherer, D.; Lorenzo Bermejo, J. Optimal selection of genetic variants for adjustment of population stratification in European association studies. Brief. Bioinform. 2019, 21, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Hellwege, J.N.; Keaton, J.M.; Giri, A.; Gao, X.; Velez Edwards, D.R.; Edwards, T.L. Population stratification in genetic association studies. Curr. Protoc. Hum. Genet. 2017, 95, 1.22.2–1.22.23. [Google Scholar] [CrossRef]
- Lacour, A.; Schuller, V.; Drichel, D.; Herold, C.; Jessen, F.; Leber, M.; Maier, W.; Noethen, M.M.; Ramirez, A.; Vaitsiakhovich, T.; et al. Novel genetic matching methods for handling population stratification in genome-wide association studies. BMC Bioinform. 2015, 16, 84. [Google Scholar] [CrossRef]
- Guan, W.; Liang, L.; Boehnke, M.; Abecasis, G.R. Genotype-based matching to correct for population stratification in large-scale case-control genetic association studies. Genet. Epidemiol. 2009, 33, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, S.J.; He, J.Q.; Burkett, K.M.; Ruan, J.; Opushnyev, I.V.; Tripp, B.W.; Zeznik, J.A.; Abara, C.O.; Nelson, C.C.; Walley, K.R. Microarray genotyping resource to determine population stratification in genetic association studies of complex disease. BioTechniques 2004, 37, 977–985. [Google Scholar] [CrossRef]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Venema, W.T.U.; Voskuil, M.D.; Vila, A.V.; Graham, D.B.; Weersma, R.K.; Festen, E.A. Single cell RNA sequencing of t cells in Crohn’s disease identifies tissue specific drug targets. Gastroenterology 2018, 154, S189. [Google Scholar] [CrossRef]
- Schmidt, F.; Efferth, T. Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals 2016, 9, 33. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Wu, S. Single-cell sequencing for drug discovery and drug development. Curr. Top. Med. Chem. 2017, 17, 1769–1777. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Name | Cancer Type | Target |
|---|---|---|
| ALN-VSP | Liver Cancer and metastases | VEGF gene, kinesin spindle (KSP) [30,31] |
| APN401 1 | Various solid tumors | CBLB protein [32] |
| Atu-027 | Advanced Solid Tumors/Pancreatic cancer | Protein Kinase N3 gene [30,31] |
| CALAA-01 | Solid Tumors | M2 subunit of ribonucleotide reductase (R2) [31,32] |
| DCR-MYC | Liver cancer, Multiple myeloma, Non-Hodgkin’s lymphoma, Pancreatic cancer, Solid tumors | C-Myc [31] |
| EphA2 | Advanced or Recurrent Solid Tumors | EphA2 [31,32,33] |
| siG12D LODER | Advanced pancreatic cancer | KRAS G12D [31,32,33] |
| SPC2996 | Chronic Lymphocytic Leukemia | Bcl-2 gene [30] |
| TKM 080301 | Neuroendocrine tumors (NET) and adrenocortical carcinoma (ACC) tumors | Pololike kinase 1 (PLK1) [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebert, M.; Jaśkiewicz, M.; Moszyńska, A.; Collawn, J.F.; Bartoszewski, R. The Effects of Single Nucleotide Polymorphisms in Cancer RNAi Therapies. Cancers 2020, 12, 3119. https://doi.org/10.3390/cancers12113119
Gebert M, Jaśkiewicz M, Moszyńska A, Collawn JF, Bartoszewski R. The Effects of Single Nucleotide Polymorphisms in Cancer RNAi Therapies. Cancers. 2020; 12(11):3119. https://doi.org/10.3390/cancers12113119
Chicago/Turabian StyleGebert, Magdalena, Maciej Jaśkiewicz, Adrianna Moszyńska, James F. Collawn, and Rafał Bartoszewski. 2020. "The Effects of Single Nucleotide Polymorphisms in Cancer RNAi Therapies" Cancers 12, no. 11: 3119. https://doi.org/10.3390/cancers12113119
APA StyleGebert, M., Jaśkiewicz, M., Moszyńska, A., Collawn, J. F., & Bartoszewski, R. (2020). The Effects of Single Nucleotide Polymorphisms in Cancer RNAi Therapies. Cancers, 12(11), 3119. https://doi.org/10.3390/cancers12113119