Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance

 , and
, and 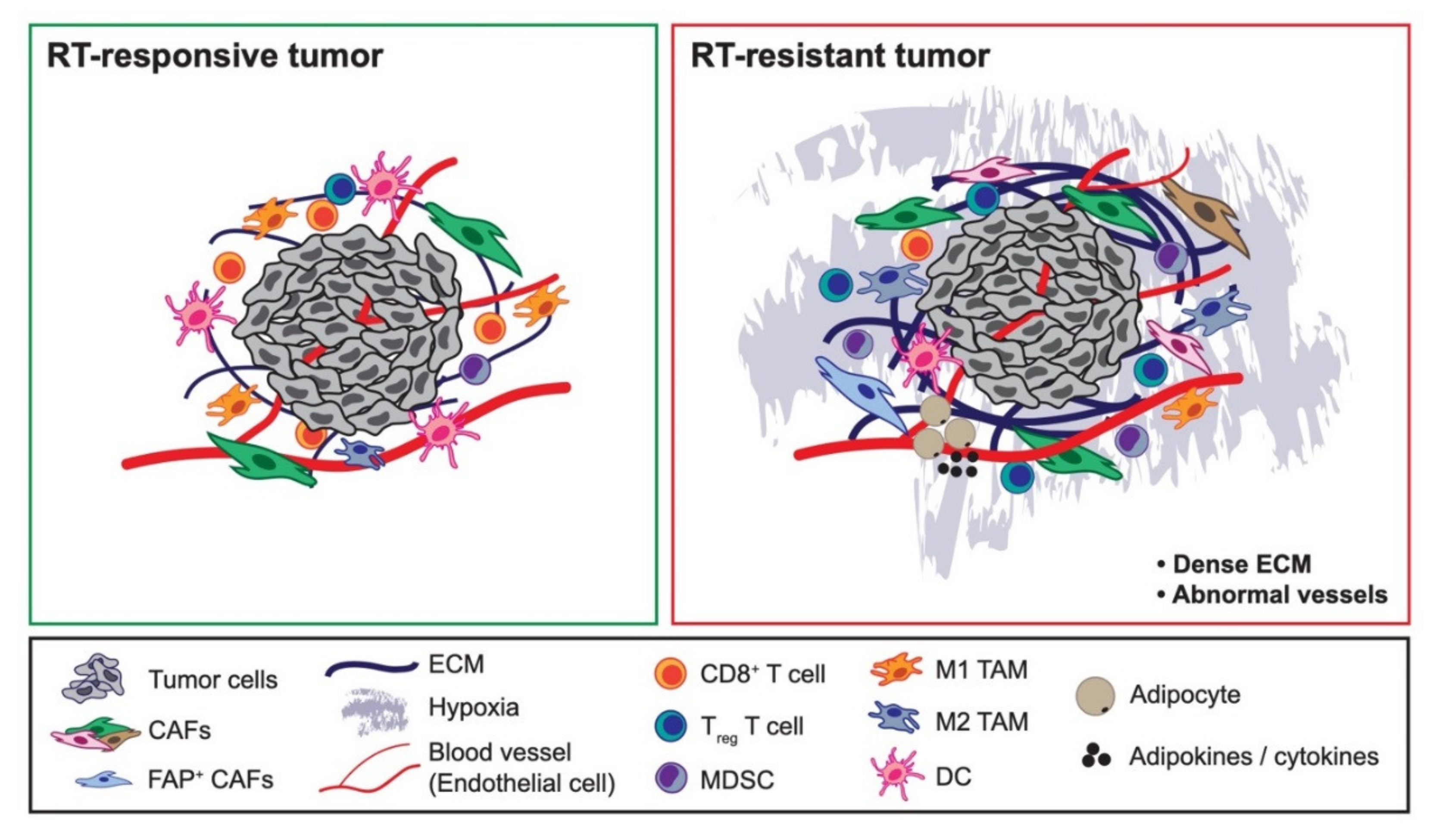{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Impact of RT on the Tumor Stroma
3. The Impact of CAFs on RT Efficacy
4. The Impact of ECM on RT Efficacy
5. The Impact of Immune Cells on RT Efficacy
6. Immunomodulatory Roles of CAFs and the ECM and How Their Interactions Regulate RT Efficacy
7. The Impact of Other Stromal Cells on RT Efficacy
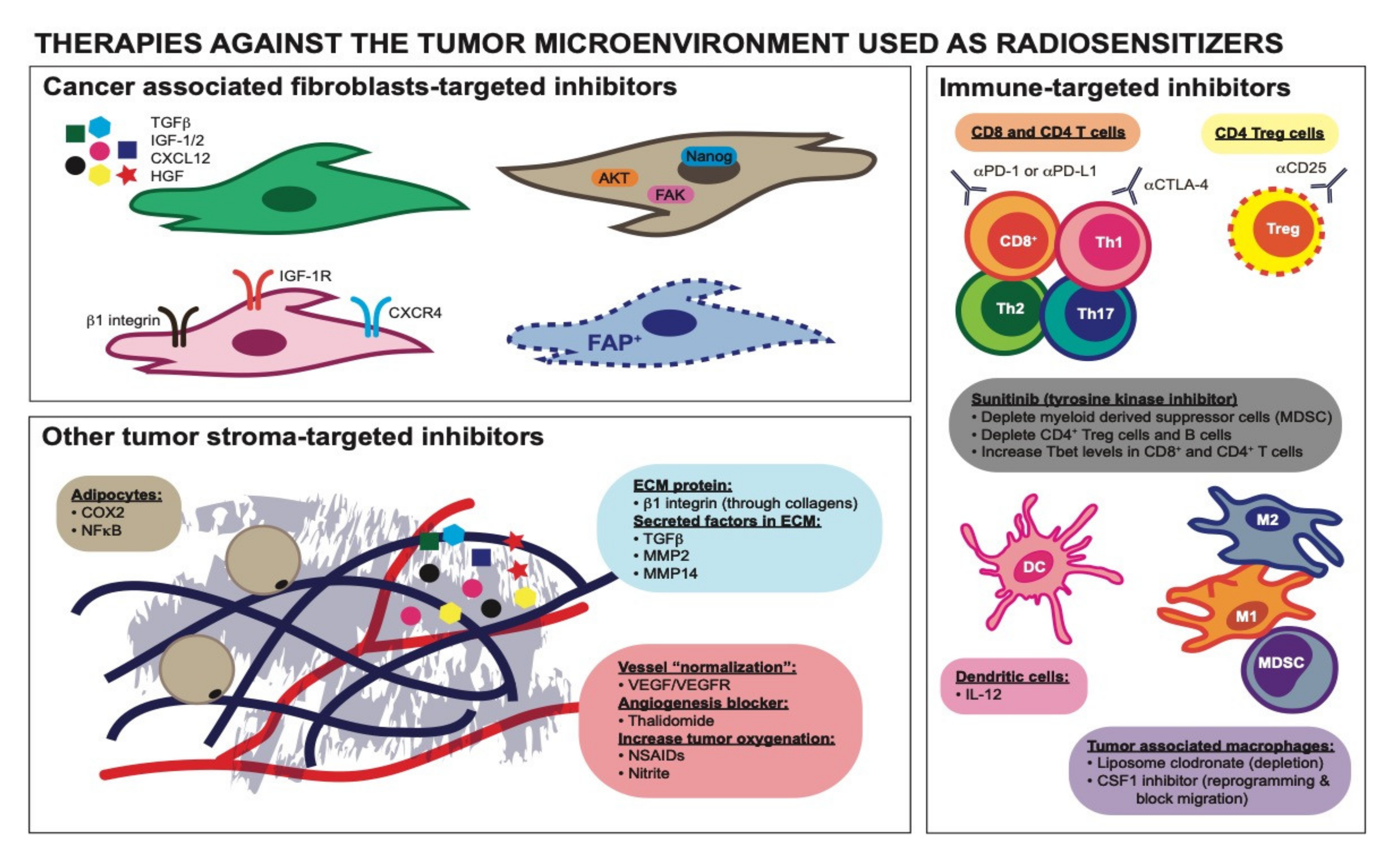
8. Forward Looking Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Junttila, M.R.; De Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Iyer, S.P.; Hunt, C.R.; Pandita, T.K. Cross Talk between Radiation and Immunotherapy: The Twain Shall Meet. Radiat. Res. 2018, 189, 219–224. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment—Tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 2018, 15, 477–494. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef]
- Grassberger, C.; Ellsworth, S.G.; Wilks, M.Q.; Keane, F.K.; Loeffler, J.S. Assessing the interactions between radiotherapy and antitumour immunity. Nat. Rev. Clin. Oncol. 2019, 16, 729–745. [Google Scholar] [CrossRef]
- Van Leeuwen, C.M.; Oei, A.L.; Crezee, J.; Bel, A.; Franken, N.A.P.; Stalpers, L.J.A.; Kok, H.P. The alfa and beta of tumours: A review of parameters of the linear-quadratic model, derived from clinical radiotherapy studies. Radiat. Oncol. 2018, 13, 96. [Google Scholar] [CrossRef]
- Hall, E.; Giaccia, A. Cell survival curves. In Radiobiology for the Radiologist, 7th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; pp. 35–53. [Google Scholar]
- Papiez, L.; Timmerman, R. Hypofractionation in radiation therapy and its impact. Med. Phys. 2008, 35, 112–118. [Google Scholar] [CrossRef]
- Mahajan, A.; Ahmed, S.; McAleer, M.F.; Weinberg, J.S.; Li, J.; Brown, P.; Settle, S.; Prabhu, S.S.; Lang, F.F.; Levine, N.; et al. Post-operative stereotactic radiosurgery versus observation for completely resected brain metastases: A single-centre, randomised, controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1040–1048. [Google Scholar] [CrossRef]
- Qiu, H.; Moravan, M.J.; Milano, M.T.; Usuki, K.Y.; Katz, A.W. SBRT for Hepatocellular Carcinoma: 8-Year Experience from a Regional Transplant Center. J. Gastrointest. Cancer 2018, 49, 463–469. [Google Scholar] [CrossRef]
- Kishan, A.U.; King, C.R. Stereotactic Body Radiotherapy for Low- and Intermediate-Risk Prostate Cancer. Semin. Radiat. Oncol. 2017, 27, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Aridgides, P.; Bogart, J. Stereotactic Body Radiation Therapy for Stage I Non-Small Cell Lung Cancer. Thorac. Surg. Clin. 2016, 26, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Mole, R.H. Whole body irradiation; radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Leroi, N.; Lallemand, F.; Coucke, P.; Noel, A.; Martinive, P. Impacts of ionizing radiation on the different compartments of the tumor microenvironment. Front. Pharmacol. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef]
- Arnold, K.M.; Flynn, N.J.; Raben, A.; Romak, L.; Yu, Y.; Dicker, A.P.; Mourtada, F.; Sims-Mourtada, J. The Impact of Radiation on the Tumor Microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Penney, D.P.; Wayne, A.; Rosenkrans, J. Cell-Cell Matrix Interactions in Induced Lung Injury: I. The Effects of X-Irradiation on Basal Laminar Proteoglycans. Radiat. Res. 1984, 99, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H. Radiation-induced Transforming Growth Factor β and Subsequent Extracellular Matrix Reorganization in Murine Mammary Gland. Cancer Res. 1993, 53, 3880–3886. [Google Scholar] [PubMed]
- Remy, J.; Wegrowski, J.; Crechet, F.; Martin, M.; Daburon, F. Long-Term Overproduction of Collagen in Radiation-Induced Fibrosis. Radiat. Res. 1991, 125, 14–19. [Google Scholar] [CrossRef]
- Chargari, C.; Clemenson, C.; Martins, I.; Perfettini, J.-L.; Deutsch, E. Understanding the functions of tumor stroma in resistance to ionizing radiation: Emerging targets for pharmacological modulation. Drug Resist. Updat. 2013, 16, 10–21. [Google Scholar] [CrossRef]
- Kamochi, N.; Nakashima, M.; Aoki, S.; Uchihashi, K.; Sugihara, H.; Toda, S.; Kudo, S. Irradiated fibroblast-induced bystander effects on invasive growth of squamous cell carcinoma under cancer-stromal cell interaction. Cancer Sci. 2008, 99, 2417–2427. [Google Scholar] [CrossRef]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to Stromal Fibroblasts Increases Invasiveness of Pancreatic Cancer Cells through Tumor-Stromal Interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef]
- Tommelein, J.; Verset, L.; Boterberg, T.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-Associated Fibroblasts Connect Metastasis-Promoting Communication in Colorectal Cancer. Front. Oncol. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gan, G.; Wang, B.; Wu, J.; Cao, Y.; Zhu, D.; Xu, Y.; Wang, X.; Han, H.; Li, X.; et al. Cancer-associated Fibroblasts Promote Irradiated Cancer Cell Recovery Through Autophagy. EBioMedicine 2017, 17, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Cordes, N. Integrin-mediated cell-matrix interactions for prosurvival and antiapoptotic signaling after genotoxic injury. Cancer Lett. 2006, 242, 11–19. [Google Scholar] [CrossRef]
- Mantoni, T.S.; Lunardi, S.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Res. 2011, 71, 3453–3458. [Google Scholar] [CrossRef]
- Al-Assar, O.; Demiciorglu, F.; Lunardi, S.; Gaspar-Carvalho, M.M.; McKenna, W.G.; Muschel, R.M.; Brunner, T.B. Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother. Oncol. 2014, 111, 243–251. [Google Scholar] [CrossRef]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Louisa Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Sampetrean, O.; Shimizu, T.; Saga, I.; Onishi, N.; Sugihara, E.; Okubo, J.; Fujita, S.; Takano, S.; Matsumura, A.; et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013, 31, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Hawsawi, N.M.; Ghebeh, H.; Hendrayani, S.F.; Tulbah, A.; Al-Eid, M.; Al-Tweigeri, T.; Ajarim, D.; Alaiya, A.; Dermime, S.; Aboussekhra, A. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 2008, 68, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int. J. Radiat. Biol. 2015, 91, 13–27. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Tsai, K.K.C.; Stuart, J.; Chuang, Y.-Y.E.; Little, J.B.; Yuan, Z.-M. Low-Dose Radiation-Induced Senescent Stromal Fibroblasts Render Nearby Breast Cancer Cells Radioresistant. Radiat. Res. 2009, 172, 306–313. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence- associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef]
- Li, D.; Qu, C.; Ning, Z.; Wang, H.; Zang, K.; Zhuang, L.; Chen, L.; Wang, P.; Meng, Z. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 2016, 6, 2192–2206. [Google Scholar]
- Hellevik, T.; Pettersen, I.; Berg, V.; Winberg, J.O.; Moe, B.T.; Bartnes, K.; Paulssen, R.H.; Busund, L.T.; Bremnes, R.; Chalmers, A.; et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat. Oncol. 2012, 7. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Puthawala, K.; Hadjiangelis, N.; Jacoby, S.C.; Bayongan, E.; Zhao, Z.; Yang, Z.; Devitt, M.L.; Horan, G.S.; Weinreb, P.H.; Lukashev, M.E.; et al. Inhibition of integrin αvβ6, an activator of latent transforming growth factor-β, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 82–90. [Google Scholar] [CrossRef]
- Park, C.C.; Zhang, H.; Pallavicini, M.; Gray, J.W.; Baehner, F.; Park, C.J.; Bissell, M.J. Β1 Integrin Inhibitory Antibody Induces Apoptosis of Breast Cancer Cells, Inhibits Growth, and Distinguishes Malignant from Normal Phenotype in Three Dimensional Cultures and in Vivo. Cancer Res. 2006, 66, 1526–1535. [Google Scholar] [CrossRef]
- Park, C.C.; Zhang, H.J.; Yao, E.S.; Park, C.J.; Bissell, M.J. Β1 Integrin Inhibition Dramatically Enhances Radiotherapy Efficacy in Human Breast Cancer Xenografts. Cancer Res. 2008, 68, 4398–4405. [Google Scholar] [CrossRef] [PubMed]
- Cordes, N.; Seidler, J.; Durzok, R.; Geinitz, H.; Brakebusch, C. Β1-Integrin-Mediated Signaling Essentially Contributes to Cell Survival After Radiation-Induced Genotoxic Injury. Oncogene 2006, 25, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Freeman, M.L.; Arteaga, C.L.; Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; et al. Inhibition of TGF Beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression Find the latest version: Inhibition of TGF- β with neutralizing antibodies prevents radiation-induced acceleration of metastatic. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Hellevik, T.; Martinez-Zubiaurre, I. Radiotherapy and the tumor stroma: The importance of dose and fractionation. Front. Oncol. 2014, 4, 1–12. [Google Scholar] [CrossRef]
- Chetty, C.; Bhoopathi, P.; Rao, J.S.; Lakka, S.S. Inhibition of matrix metalloproteinase-2 enhances radiosensitivity by abrogating radiation-induced FoxM1-mediated G2/M arrest in A549 lung cancer cells. Int. J. Cancer 2009, 124, 2468–2477. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The Concentration of Oxygen Dissolved in Tissues at the Time of Irradiation as a Factor in Radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Brown, J.M. The hypoxic cell: A target for selective cancer therapy—Eighteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar]
- Brown, J.M. Vasculogenesis: A crucial player in the resistance of solid tumours to radiotherapy. Br. J. Radiol. 2014, 87. [Google Scholar] [CrossRef]
- Frérart, F.; Sonveaux, P.; Rath, G.; Smoos, A.; Meqor, A.; Charlier, N.; Jordan, B.F.; Saliez, J.; Noel, A.; Dessy, C.; et al. The acidic tumor microenvironment promotes the reconversion of nitrite into nitric oxide: Towards a new and safe radiosensitizing strategy. Clin. Cancer Res. 2008, 14, 2768–2774. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Wink, D.A.; DeGraff, W.; Gamson, J.; Keefer, L.K.; Krishna, M.C. Hypoxic mammalian cell radiosensitization by nitric oxide. Cancer Res. 1993, 53, 5845–5848. [Google Scholar] [PubMed]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.-S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ansiaux, R.; Baudelet, C.; Jordan, B.F.; Beghein, N.; Sonveaux, P.; De Wever, J.; Martinive, P.; Grégoire, V.; Feron, O.; Gallez, B. Thalidomide radiosensitizes tumors through early changes in the tumor microenvironment. Clin. Cancer Res. 2005, 11, 743–750. [Google Scholar]
- Crokart, N.; Radermacher, K.; Jordan, B.F.; Baudelet, C.; Cron, G.O.; Grégoire, V.; Beghein, N.; Bouzin, C.; Feron, O.; Gallez, B. Tumor radiosensitization by antiinflammatory drugs: Evidence for a new mechanism involving the oxygen effect. Cancer Res. 2005, 65, 7911–7916. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Formenti, S.C. Radiation as an immunological adjuvant: Current evidence on dose and fractionation. Front. Oncol. 2012, 2, 1–7. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 3, e28518. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Blair, T.C.; Bambina, S.; Alice, A.F.; Kramer, F.; Medler, T.R.; Baird, J.R.; Broz, M.L.; Tormoen, G.W.; Troesch, V.; Crittenden, M.R.; et al. Dendritic Cell Maturation Defines Immunological Responsiveness of Tumors to Radiation Therapy. J. Immunol. 2020, 204. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; van den Broek, M. Radiotherapy Promotes Tumor-Specific Effector CD8 + T Cells via Dendritic Cell Activation. J. Immunol. 2012, 189, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.A.; Sedlacek, A.L.; Cron, K.R.; Murphy, S.P.; Frelinger, J.G.; Lord, E.M. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol. 2013, 182, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Filatenkov, A.; Baker, J.; Mueller, A.M.S.; Kenkel, J.; Ahn, G.O.; Dutt, S.; Zhang, N.; Kohrt, H.; Jensen, K.; Dejbakhsh-Jones, S.; et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin. Cancer Res. 2015, 21, 3727–3739. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol. Res. 2015, 3, 345–355. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Suzuki, Y.; Mimura, K.; Ando, K.; Oike, T.; Sato, H.; Okonogi, N.; Maruyama, T.; Izawa, S.; Noda, S.E.; et al. Radiotherapy-induced anti-tumor immunity contributes to the therapeutic efficacy of irradiation and can be augmented by CTLA-4 blockade in a mouse model. PLoS ONE 2014, 9, e92572. [Google Scholar] [CrossRef]
- Gerber, S.A.; Lim, J.Y.H.; Connolly, K.A.; Sedlacek, A.L.; Barlow, M.L.; Murphy, S.P.; Egilmez, N.K.; Lord, E.M. Radio-responsive tumors exhibit greater intratumoral immune activity than nonresponsive tumors. Int. J. Cancer 2014, 134, 2383–2392. [Google Scholar] [CrossRef][Green Version]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Park, S.S.; Dong, H.; Liu, X.; Harrington, S.M.; Krco, C.J.; Grams, M.P.; Mansfield, A.S.; Furutani, K.M.; Olivier, K.R.; Kwon, E.D. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol. Res. 2015, 3, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Milas, L. Tumor Bed Effect in Murine Tumors: Relationship to Tumor Take and Tumor Macrophage Content. Radiat. Res. 1990, 123, 232. [Google Scholar] [CrossRef] [PubMed]
- Vatner, R.E.; Formenti, S.C. Myeloid-derived cells in tumors: Effects of radiation. Semin. Radiat. Oncol. 2015, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Meng, Y.; Beckett, M.A.; Liang, H.; Mauceri, H.J.; Van Rooijen, N.; Cohen, K.S.; Weichselbaum, R.R. Blockade of tumor necrosis factor α signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010, 70, 1534–1543. [Google Scholar] [CrossRef]
- Prakash, H.; Klug, F.; Nadella, V.; Mazumdar, V.; Schmitz-Winnenthal, H.; Umansky, L. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: Lesson from insulinoma. Carcinogenesis 2016, 37, 301–313. [Google Scholar] [CrossRef]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS⁺/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.S.; Fu, S.Y.; Wang, S.C.; Yu, C.F.; Chen, F.H.; Lin, C.M.; Hong, J.H. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Kioi, M.; Nakashima, H.; Sugiura, K.; Mitsudo, K.; Aoki, I.; Taniguchi, H.; Tohnai, I. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci. Rep. 2016, 6, 27548. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Nguy, S.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Daley, D.; Barilla, R.; et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016, 150, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, M.R.; Cottam, B.; Savage, T.; Nguyen, C.; Newell, P.; Gough, M.J. Expression of NF-κb p50 in tumor stroma limits the control of tumors by radiation therapy. PLoS ONE 2012, 7, e39295. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-polarized CD4+ T Cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef]
- Chen, H.M.; Ma, G.; Gildener-Leapman, N.; Eisenstein, S.; Coakley, B.A.; Ozao, J.; Mandeli, J.; Divino, C.; Schwartz, M.; Sung, M.; et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin. Cancer Res. 2015, 21, 4073–4085. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef]
- Persa, E.; Balogh, A.; Sáfrány, G.; Lumniczky, K. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. 2015, 368, 252–261. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-Regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.P.; Demarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Qinfeng, S.; Depu, W.; Xiaofeng, Y.; Shah, W.; Hongwei, C.; Yili, W. In situ observation of the effects of local irradiation on cytotoxic and regulatory T lymphocytes in cervical cancer tissue. Radiat. Res. 2013, 179, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Hori, S. Full restoration of peripheral Foxp3+ regulatory T cell pool by radioresistant host cells in scurfy bone marrow chimeras. Proc. Natl. Acad. Sci. USA 2007, 104, 8959–8964. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; Xie, M.W.; Ratikan, J.A.; McBride, W.H. Regulatory T cells in radiotherapeutic responses. Front. Oncol. 2012, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.J.; Darragh, L.; Phan, A.; Binder, D.; Bhatia, S.; Mueller, A.; Court, B.V.; Milner, D.; Raben, D.; Woessner, R.; et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J. Natl. Cancer Inst. 2019, 111, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Oweida, A.; Lennon, S.; Darragh, L.B.; Milner, D.; Phan, A.V.; Mueller, A.C.; Van Court, B.; Raben, D.; Serkova, N.J.; et al. Inhibition of EphB4-Ephrin-B2 Signaling Reprograms the Tumor Immune Microenvironment in Head and Neck Cancers. Cancer Res. 2019, 79, 2722–2735. [Google Scholar] [CrossRef]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing tumor immunity with fractionated radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Bastian, A.; Morin, S.; Marte, J.; Beetham, P.; Tsang, K.Y.; Yokokawa, J.; Hodge, J.W.; Ménard, C.; et al. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin. Cancer Res. 2005, 11, 3353–3362. [Google Scholar] [CrossRef]
- Schaue, D.; Comin-Anduix, B.; Ribas, A.; Zhang, L.; Goodglick, L.; Sayre, J.W.; Debucquoy, A.; Haustermans, K.; McBride, W.H. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin. Cancer Res. 2008, 14, 4883–4890. [Google Scholar] [CrossRef]
- Rutkowski, J.; Ślebioda, T.; Kmieć, Z.; Zaucha, R. Changes in systemic immune response after stereotactic ablative radiotherapy. Preliminary results of a prospective study in patients with early lung cancer. Pol. Arch. Intern. Med. 2017, 127, 245–253. [Google Scholar] [CrossRef]
- Wasserman, J.; Blomgren, H.; Rotstein, S.; Petrini, B.; Hammarström, S. Immunosuppression in irradiated breast cancer patients: In vitro effect of cyclooxygenase inhibitors. Bull. N. Y. Acad. Med. 1989, 65, 36–44. [Google Scholar]
- Rödel, F.; Frey, B.; Manda, K.; Hildebrandt, G.; Hehlgans, S.; Keilholz, L.; Seegenschmiedt, M.H.; Gaipl, U.S.; Rödel, C. Immunomodulatory properties and molecular effects in inflammatory diseases of low-dose x-irradiation. Front. Oncol. 2012, 2, 120. [Google Scholar] [CrossRef]
- Trowell, O.A. The sensitivity of lymphocytes to ionising radiation. J. Pathol. Bacteriol. 1952, 64, 687–704. [Google Scholar] [CrossRef]
- Frey, B.; Rückert, M.; Weber, J.; Mayr, X.; Derer, A.; Lotter, M.; Bert, C.; Rödel, F.; Fietkau, R.; Gaipl, U.S. Hypofractionated Irradiation Has Immune Stimulatory Potential and Induces a Timely Restricted Infiltration of Immune Cells in Colon Cancer Tumors. Front. Immunol. 2017, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Demaria, S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat. Res. 2010, 173, 418–425. [Google Scholar] [CrossRef]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol. 2008, 180, 3132–3139. [Google Scholar] [CrossRef]
- Hallahan, D.E.; Spriggs, D.R.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc. Natl. Acad. Sci. USA 1989, 86, 10104–10107. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Derynck, R.; Tsang, M.L.; Weatherbee, J.A. Transforming growth factor-beta activation in irradiated murine mammary gland. J. Clin. Investig. 1994, 93, 892–899. [Google Scholar] [CrossRef]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [CrossRef]
- Klein, B.; Loven, D.; Lurie, H.; Rakowsky, E.; Nyska, A.; Levin, I.; Klein, T. The effect of irradiation on expression of HLA class I antigens in human brain tumors in culture. J. Neurosurg. 1994, 80, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Hiserodt, J.C.; Chiriva-Internati, M.; Woodliff, J.; Theus, J.W.; Barclay, D.; Pecorelli, S.; Parham, G.P. Effects of irradiation on the expression of major histocompatibility complex class I antigen and adhesion costimulation molecules ICAM-1 in human cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 1997, 39, 737–742. [Google Scholar] [CrossRef]
- Santin, A.D.; Hiserodt, J.C.; Fruehauf, J.; DiSaia, P.J.; Pecorelli, S.; Granger, G.A. Effects of irradiation on the expression of surface antigens in human ovarian cancer. Gynecol. Oncol. 1996, 60, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Lourdes Mora-García, M.; García-Rocha, R.; Morales-Ramírez, O.; Montesinos, J.J.; Weiss-Steider, B.; Hernández-Montes, J.; Ávila-Ibarra, L.R.; Don-López, C.A.; Velasco-Velázquez, M.A.; Gutiérrez-Serrano, V.; et al. Mesenchymal stromal cells derived from cervical cancer produce high amounts of adenosine to suppress cytotoxic T lymphocyte functions. J. Transl. Med. 2016, 14, 1–14. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.t.; Deng, Y.n.; Yi, H.m.; Wang, G.y.; Fu, B.s.; Chen, W.j.; Liu, W.; Tai, Y.; Peng, Y.w.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef]
- Khosravi-Maharlooei, M.; Pakyari, M.; Jalili, R.B.; Salimi-Elizei, S.; Lai, J.C.Y.; Poormasjedi-Meibod, M.; Kilani, R.T.; Dutz, J.; Ghahary, A. Tolerogenic effect of mouse fibroblasts on dendritic cells. Immunology 2016, 148, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res. 2013, 73, 3007. [Google Scholar] [CrossRef]
- Kim, J.H.; Oh, S.-H.; Kim, E.-J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; Kim, T.I.; Kim, W.H. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochem. Biophys. Res. Commun. 2012, 423, 60–66. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. PNAS 2013, 110, 20212. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- Ager, E.I.; Kozin, S.V.; Kirkpatrick, N.D.; Seano, G.; Kodack, D.P.; Askoxylakis, V.; Huang, Y.; Goel, S.; Snuderl, M.; Muzikansky, A.; et al. Blockade of MMP14 activity in murine breast carcinomas: Implications for macrophages, vessels, and radiotherapy. J. Natl. Cancer Inst. 2015, 107, 1–12. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kibria, G.; Hatakeyama, H.; Harashima, H. Cancer multidrug resistance: Mechanisms involved and strategies for circumvention using a drug delivery system. Arch. Pharm. Res. 2014, 37, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Griffin, R.J.; Hui, S.; Levitt, S.H.; Song, C.W. Radiation-induced vascular damage in tumors: Implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat. Res. 2012, 177, 311–327. [Google Scholar] [CrossRef]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef]
- Kocher, M.; Treuer, H.; Voges, J.; Hoevels, M.; Sturm, V.; Müller, R.P. Computer simulation of cytotoxic and vascular effects of radiosurgery in solid and necrotic brain metastases. Radiother. Oncol. 2000, 54, 149–156. [Google Scholar] [CrossRef]
- Sonveaux, P.; Dessy, C.; Brouet, A.; Jordan, B.F.; Grégoire, V.; Gallez, B.; Balligand, J.L.; Feron, O. Modulation of the tumor vasculature functionality by ionizing radiation accounts for tumor radiosensitization and promotes gene delivery. FASEB J. 2002, 16, 1979–1981. [Google Scholar] [CrossRef]
- Crokart, N.; Jordan, B.F.; Baudelet, C.; Ansiaux, R.; Sonveaux, P.; Grégoire, V.; Beghein, N.; DeWever, J.; Bouzin, C.; Feron, O.; et al. Early reoxygenation in tumors after irradiation: Determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 901–910. [Google Scholar] [CrossRef]
- Rao, S.S.; Thompson, C.; Cheng, J.; Haimovitz-Friedman, A.; Powell, S.N.; Fuks, Z.; Kolesnick, R.N. Axitinib sensitization of high Single Dose Radiotherapy. Radiother. Oncol. 2014, 111, 88–93. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun 2011, 411, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Tang, X.; Yang, Z.; Benesch, M.G.K.; Marshall, A.; Murray, D.; Hemmings, D.G.; Wuest, F.; McMullen, T.P.W.; Brindley, D.N. Implications for breast cancer treatment from increased autotaxin production in adipose tissue after radiotherapy. FASEB J. 2017, 31, 4064–4077. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Vozenin, M.C.; Hendry, J.H.; Limoli, C.L. Biological Benefits of Ultra-high Dose Rate FLASH Radiotherapy: Sleeping Beauty Awoken. Clin. Oncol. 2019, 31, 407–415. [Google Scholar] [CrossRef]
- Harrington, K.J. Ultrahigh dose-rate radiotherapy: Next steps for FLASH-RT. Clin. Cancer Res. 2019, 25, 3–5. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krisnawan, V.E.; Stanley, J.A.; Schwarz, J.K.; DeNardo, D.G. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers 2020, 12, 2916. https://doi.org/10.3390/cancers12102916
Krisnawan VE, Stanley JA, Schwarz JK, DeNardo DG. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers. 2020; 12(10):2916. https://doi.org/10.3390/cancers12102916
Chicago/Turabian StyleKrisnawan, Varintra E., Jennifer A. Stanley, Julie K. Schwarz, and David G. DeNardo. 2020. "Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance" Cancers 12, no. 10: 2916. https://doi.org/10.3390/cancers12102916
APA StyleKrisnawan, V. E., Stanley, J. A., Schwarz, J. K., & DeNardo, D. G. (2020). Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers, 12(10), 2916. https://doi.org/10.3390/cancers12102916