Understanding the Effects of Radiotherapy on the Tumour Immune Microenvironment to Identify Potential Prognostic and Predictive Biomarkers of Radiotherapy Response



Simple Summary
Abstract
1. Introduction
2. The Tumour Immune Microenvironment
2.1. The Components and Classification of TME
2.2. The Effects of RT on the Tumour Microenvironment
2.2.1. RT-Induced Immunostimulation
2.2.2. RT-Induced Immunosuppression
2.2.3. RT and Cell Death
3. Biomarkers
3.1. Radiotherapy-Related Biomarkers
3.1.1. PD-1/PD-L1 Expression
3.1.2. Immune Infiltrates
3.1.3. Immunoscore
3.1.4. Gene Expression Profiling
3.1.5. Neoantigens
3.2. Imaging Biomarkers
3.3. Biomarkers for RT and ICI Combination Therapies
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Herrera, F.G.; Bourhis, J.; Coukos, G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA A Cancer J. Clin. 2016, 67, 65–85. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Ponzetta, A.; Inforzato, A.; Jaillon, S. Innate immunity, inflammation and tumour progression: Double-edged swords. J. Intern. Med. 2019, 285, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In Situ Cytotoxic and Memory T Cells Predict Outcome in Patients With Early-Stage Colorectal Cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Galon, J.; Pagès, F.; Marincola, F.M.; Angell, H.K.; Thurin, M.; Lugli, A.; Zlobec, I.; Berger, A.; Bifulco, C.B.; Botti, G.; et al. Cancer classification using the Immunoscore: A worldwide task force. J. Transl. Med. 2012, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunol. 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef]
- Mole, R.H. Whole Body Irradiation—Radiobiology or Medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G.; Francisca, B.J. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol. Res. 2014, 3, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Victor, C.T.-S.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; DeMaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Nesslinger, N.J.; Sahota, R.A.; Stone, B.; Johnson, K.; Chima, N.; King, C.; Rasmussen, D.; Bishop, D.; Rennie, P.S.; Gleave, M.; et al. Standard Treatments Induce Antigen-Specific Immune Responses in Prostate Cancer. Clin. Cancer Res. 2007, 13, 1493–1502. [Google Scholar] [CrossRef]
- Schaue, D.; Comin-Anduix, B.; Ribas, A.; Zhang, L.; Goodglick, L.; Sayre, J.W.; Debucquoy, A.; Haustermans, K.; McBride, W.H. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin. Cancer Res. 2008, 14, 4883–4890. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic Correlates of the Abscopal Effect in a Patient with Melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; K.Wansley, E.; Camphausen, K.; Luiten, R.M.; De Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2006, 13, 54–61. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Honeychurch, J.; Illidge, T.M. The influence of radiation in the context of developing combination immunotherapies in cancer. Ther. Adv. Vaccines Immunother. 2018, 5, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Formenti, S.C. Radiation-Induced Chromosomal Aberrations and Immunotherapy: Micronuclei, Cytosolic DNA, and Interferon-Production Pathway. Front. Oncol. 2018, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; DeMaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, X.; Wang, X.; Zhao, Z.; Hu, W.; Zeng, S.; Wei, J.; Yang, X.; Qian, L.; Zhou, S.; et al. The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 171. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Tan, Y.; Wei, Q.; Yu, W. Cancer-associated fibroblasts in radiotherapy: Challenges and new opportunities. Cell Commun. Signal. 2019, 17, 47. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Jäättelä, M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene 2004, 23, 2746–2756. [Google Scholar] [CrossRef]
- Long, J.S.; Ryan, K.M. New frontiers in promoting tumour cell death: Targeting apoptosis, necroptosis and autophagy. Oncogene 2012, 31, 5045–5060. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Monje, M.; Wen, P.Y. Chapter 15—Neurologic Complications of Oncologic Therapy. In Handbook of Neuro-Oncology Neuroimaging, 2nd ed.; Newton, H.B., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 125–142. [Google Scholar]
- Omay, S.B.; Atsina, K.K.; Baehring, J.M. Chapter 53—Nonneoplastic Mass Lesions of the Central Nervous System. In Handbook of Neuro-Oncology Neuroimaging, 2nd ed.; Newton, H.B., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 653–665. [Google Scholar]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Montani, M.S.G.; Granato, M.; Garufi, A.; Faggioni, A.; D’Orazi, G. Autophagy manipulation as a strategy for efficient anticancer therapies: Possible consequences. J. Exp. Clin. Cancer Res. 2019, 38, 262. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Mariño, G.; Kepp, O.; Michaud, M.; Perfettini, J.-L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2013, 21, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Rojas-Puentes, L.L.; González-Pinedo, M.; Crismatt, A.; Gómez, A.O.; Gamboa-Vignolle, C.; Núñez-Gómez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 2013, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Royce, G.H.; Brown-Borg, H.M.; Deepa, S.S. The potential role of necroptosis in inflammaging and aging. GeroScience 2019, 41, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, M.; He, S.; Luo, Y.; Zhao, Y.; Cheng, J.; Gong, Y.; Xie, J.; Wang, Y.; Hu, B.; et al. Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Gutman, S.; Kessler, L.G. The US Food and Drug Administration perspective on cancer biomarker development. Nat. Rev. Cancer 2006, 6, 565–571. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Bosco, A.; Millward, M.J.; Small, M.; Nowak, A.K.; Lake, R.A. Dynamic versus static biomarkers in cancer immune checkpoint blockade: Unravelling complexity. Nat. Rev. Drug Discov. 2017, 16, 264–272. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2019, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, H.; Zeng, Q.; Guo, X.-J.; Wang, H.; Liu, H.-H.; Dong, Z.-Y. HPV-positive status associated with inflamed immune microenvironment and improved response to anti-PD-1 therapy in head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 13404. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.D.; Ross, J.A.; McLaren, D.; Parker, C.C.; Habib, F.K.; Riddick, A.C. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010, 105, 8–13. [Google Scholar] [CrossRef]
- Rothenberger, N.J.; Somasundaram, A.; Stabile, L.P. The Role of the Estrogen Pathway in the Tumor Microenvironment. Int. J. Mol. Sci. 2018, 19, 611. [Google Scholar] [CrossRef]
- Onal, C.; Yildirim, B.A.; Guler, O.C.; Mertsoylu, H. The Utility of Pretreatment and Posttreatment Lymphopenia in Cervical Squamous Cell Carcinoma Patients Treated with Definitive Chemoradiotherapy. Int. J. Gynecol. Cancer 2018, 28, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; An, G.; Xie, S.; Yao, Y.; Feng, G. The clinical and prognostic significance of CD14+HLA-DR−/low myeloid-derived suppressor cells in hepatocellular carcinoma patients receiving radiotherapy. Tumor Biol. 2016, 37, 10427–10433. [Google Scholar] [CrossRef]
- Napolitano, M.; D’Alterio, C.; Cardone, E.; Trotta, A.; Pecori, B.; Rega, D.; Pace, U.; Scala, D.; Scognamiglio, G.; Tatangelo, F.; et al. Peripheral myeloid-derived suppressor and T regulatory PD-1 positive cells predict response to neoadjuvant short-course radiotherapy in rectal cancer patients. Oncotarget 2015, 6, 8261–8270. [Google Scholar] [CrossRef]
- Willett, C.G.; Duda, D.G.; Di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, Safety, and Biomarkers of Neoadjuvant Bevacizumab, Radiation Therapy, and Fluorouracil in Rectal Cancer: A Multidisciplinary Phase II Study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef]
- Balermpas, P.; Michel, Y.; Wagenblast, J.; Seitz, O.; Weiss, C.; Rödel, F.; Rödel, C.; Fokas, E. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br. J. Cancer 2013, 110, 501–509. [Google Scholar] [CrossRef]
- Anitei, M.-G.; Zeitoun, G.; Mlecnik, B.; Marliot, F.; Haicheur, N.; Todosi, A.M.; Kirilovsky, A.; Lagorce, C.; Bindea, G.; Ferariu, D.; et al. Prognostic and Predictive Values of the Immunoscore in Patients with Rectal Cancer. Clin. Cancer Res. 2014, 20, 1891–1899. [Google Scholar] [CrossRef]
- Lim, Y.J.; Koh, J.; Kim, K.; Chie, E.K.; Kim, B.; Lee, K.; Jang, J.-Y.; Kim, S.-W.; Oh, -Y.; Bang, Y.-J.; et al. High ratio of programmed cell death protein 1 (PD-1)+/CD8+ tumor-infiltrating lymphocytes identifies a poor prognostic subset of extrahepatic bile duct cancer undergoing surgery plus adjuvant chemoradiotherapy. Radiother. Oncol. 2015, 117, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Okonogi, N.; Nakajima, N.I.; Morokoshi, Y.; Kanda, H.; Yamada, T.; Kobayashi, Y.; Banno, K.; Wakatsuki, M.; Yamada, S.; et al. Significance of PD-L1 expression in carbon-ion radiotherapy for uterine cervical adeno/adenosquamous carcinoma. J. Gynecol. Oncol. 2020, 31, 19. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, S.; Kim, K.-J.; Seong, J. Clinical significance of soluble programmed cell death ligand-1 (sPD-L1) in hepatocellular carcinoma patients treated with radiotherapy. Radiother. Oncol. 2018, 129, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.T.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; De Andrade, L.F.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Eder, T.; Hess, A.; Konschak, R.; Stromberger, C.; Jöhrens, K.; Fleischer, V.; Hummel, M.; Balermpas, P.; Von Der Grün, J.; Linge, A.; et al. Interference of tumour mutational burden with outcome of patients with head and neck cancer treated with definitive chemoradiation: A multicentre retrospective study of the German Cancer Consortium Radiation Oncology Group. Eur. J. Cancer 2019, 116, 67–76. [Google Scholar] [CrossRef]
- Efstathiou, J.A.; Mouw, K.W.; Gibb, E.A.; Liu, Y.; Wu, C.-L.; Drumm, M.R.; Da Costa, J.B.; Du Plessis, M.; Wang, N.Q.; Davicioni, E.; et al. Impact of Immune and Stromal Infiltration on Outcomes Following Bladder-Sparing Trimodality Therapy for Muscle-Invasive Bladder Cancer. Eur. Urol. 2019, 76, 59–68. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D.; E Valsecchi, M. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Do, H.B.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, E.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.H.F.; Lo, A.W.; Leung, C.-Y.; Shek, W.-H.; Kwong, R.L.W.; Lam, K.-O.; Tong, C.-C.; Sze, C.-K.; Leung, T.-W. Correlation of PD-L1 Expression of Tumor Cells with Survival Outcomes after Radical Intensity-Modulated Radiation Therapy for Non-Metastatic Nasopharyngeal Carcinoma. PLoS ONE 2016, 11, e0157969. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Hollander, P.; Amini, R.-M.; Ginman, B.; Molin, D.; Enblad, G.; Glimelius, I. Expression of PD-1 and PD-L1 increase in consecutive biopsies in patients with classical Hodgkin lymphoma. PLoS ONE 2018, 13, e0204870. [Google Scholar] [CrossRef]
- De Re, V.; Caggiari, L.; Repetto, O.; Mussolin, L.; Mascarin, M.; Re, D. Classical Hodgkin’s Lymphoma in the Era of Immune Checkpoint Inhibition. J. Clin. Med. 2019, 8, 1596. [Google Scholar] [CrossRef]
- Qin, Q.; Nan, X.; Miller, T.; Fisher, R.; Teh, B.; Pandita, S.; Farach, A.M.; Pingali, S.R.; Pandita, R.K.; Butler, E.B.; et al. Complete Local and Abscopal Responses from a Combination of Radiation and Nivolumab in Refractory Hodgkin’s Lymphoma. Radiat. Res. 2018, 190, 322–329. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Popple, A.; Illidge, T. Harmonization of programmed cell death ligand-1 diagnostic assays in non-small cell lung cancer. Transl. Cancer Res. 2017, 6, S553–S556. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Rimm, D.L.; Han, G.; Taube, J.M.; Yi, E.S.; Bridge, J.A.; Flieder, U.B.; Homer, R.; West, W.W.; Wu, H.; Roden, A.C.; et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol. 2017, 3, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- FDA Nods for Atezolizumab and Nivolumab. Cancer Discov. 2016, 6, 811. [CrossRef] [PubMed]
- Rashidian, M.; Ingram, J.R.; Dougan, M.; Dongre, A.; Whang, K.A.; Le Gall, C.; Cragnolini, J.J.; Bierie, B.; Gostissa, M.; Gorman, J.; et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J. Exp. Med. 2017, 214, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Xiao, Y.; Peng, C.; Liu, T.; Lin, Z.; Yang, Q.; Zhang, T.; Liu, J.; Ma, H. 53BP1 expression and immunoscore are associated with the efficacy of neoadjuvant chemoradiotherapy for rectal cancer. Strahlenther. Onkol. 2019, 196, 465–473. [Google Scholar] [CrossRef]
- Teng, F.; Mu, D.; Meng, X.; Kong, L.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor infiltrating lymphocytes (TILs) before and after neoadjuvant chemoradiotherapy and its clinical utility for rectal cancer. Am. J. Cancer Res. 2015, 5, 2064–2074. [Google Scholar]
- Hagland, H.; Lea, D.; Watson, M.; Søreide, K. Correlation of circulating T-cells in pre-operative blood to intratumoral density and location of CD3+ and CD8+ T-cells in colorectal cancer: A potential for an immunoscore by liquid biopsy? Eur. J. Cancer 2017, 72, S49–S50. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Fuchs, E.; Ricken, G.; Mlecnik, B.; Bindea, G.; Spanberger, T.; Hackl, M.; Widhalm, G.; Dieckmann, K.; Prayer, D.; et al. Density of tumor-infiltrating lymphocytes correlates with extent of brain edema and overall survival time in patients with brain metastases. OncoImmunology 2015, 5, e1057388. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasić, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Grassberger, C.; Ellsworth, S.G.; Wilks, M.Q.; Keane, F.K.; Loeffler, J.S. Assessing the interactions between radiotherapy and antitumour immunity. Nat. Rev. Clin. Oncol. 2019, 16, 729–745. [Google Scholar] [CrossRef]
- Agostinelli, C.; Gallamini, A.; Stracqualursi, L.; Agati, P.; Tripodo, C.; Fuligni, F.; Sista, M.T.; Fanti, S.; Biggi, A.; Vitolo, U.; et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: A retrospective, European, multicentre cohort study. Lancet Haematol. 2016, 3, e467–e479. [Google Scholar] [CrossRef]
- Fischer, B.M.; Lassen, U.; Mortensen, J.; Larsen, S.; Loft, A.; Bertelsen, A.; Ravn, J.; Clementsen, P.; Høgholm, A.; Larsen, K.; et al. Preoperative Staging of Lung Cancer with Combined PET–CT. N. Engl. J. Med. 2009, 361, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Chiou, V.L.; Burotto, M. Pseudoprogression and Immune-Related Response in Solid Tumors. J. Clin. Oncol. 2015, 33, 3541–3543. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; DeMaria, S.; Formenti, S.C. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Aguilera, T.; Elghonaimy, E.A.; Shehade, H.; Rafat, M.; Castellini, L.; Jiang, D.; Kariolis, M.; Koong, A.C.; Le, Q.-T.; Ellies, L.G.; et al. Induced Tumor Heterogeneity Reveals Factors Informing Radiation and Immunotherapy Combinations. Clin. Cancer Res. 2020, 26, 2972–2985. [Google Scholar] [CrossRef]
- Schölch, S.; Rauber, C.; Tietz, A.; Rahbari, N.N.; Bork, U.; Schmidt, T.; Kahlert, C.; Haberkorn, U.; Tomai, M.A.; Lipson, K.E.; et al. Radiotherapy combined with TLR7/8 activation induces strong immune responses against gastrointestinal tumors. Oncotarget 2014, 6, 4663–4676. [Google Scholar] [CrossRef]
- Brøndum, L.; Eriksen, J.G.; Sørensen, B.S.; Mortensen, L.S.; Toustrup, K.; Overgaard, J.; Alsner, J. Plasma proteins as prognostic biomarkers in radiotherapy treated head and neck cancer patients. Clin. Transl. Radiat. Oncol. 2017, 2, 46–52. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.K.; Nebozhyn, M.V.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Karachaliou, N.; González-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Giménez-Capitán, A.; Teixido, C.; Molina-Vila, M.Á.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
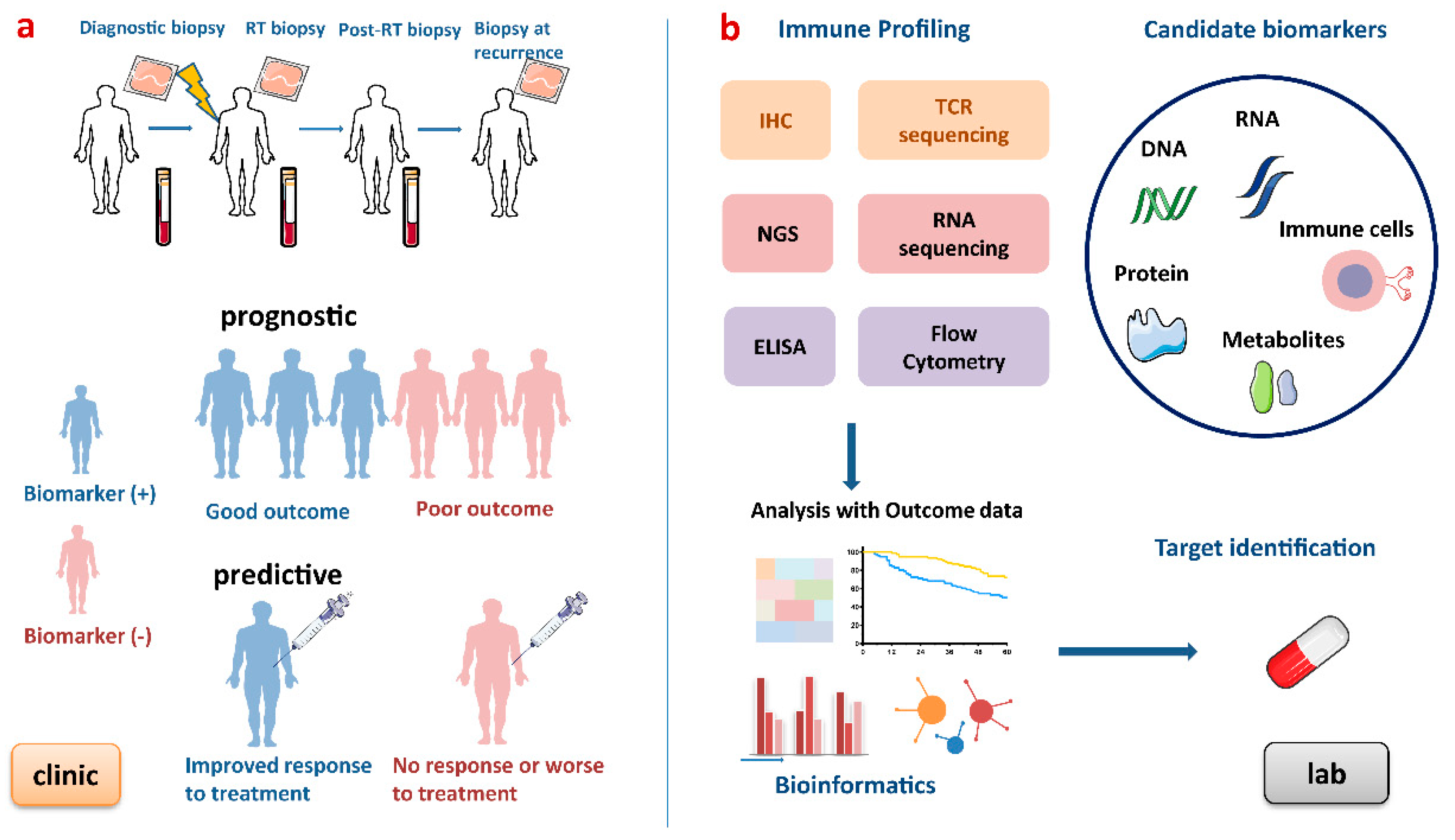{kind=link}
| Category | Sample Origin | Biomarker | Cancer Type | Treatment | Clinical Response | Reference |
|---|---|---|---|---|---|---|
| Circulating cells | Peripheral blood mononuclear cell | Lymphopenia | Cervical cancer | Definitive CRT | ↓ OS, DFS | [57] |
| Whole blood | ↑ Myeloid-derived suppressor cell | HCC | RT | ↓ OS | [58] | |
| Whole blood | ↑ MDSCs + Tregs | Rectal cancer | Short-term pCRT + surgery | Early marker of response | [59] | |
| Circulating cytokine | Plasma | VEGF, PIGF and IL-6 | CRC | Neoadjuvant bevacizumab + CRT + surgery | Predictive of response | [60] |
| Immune Infiltrates | FFPE tissue | ↑ CD3+, CD8+ TILs | HNSCC | Definitive CRT | ↑ OS, PFS Predictive of CRT | [61] |
| FFPE tissue, surgical specimens | ↑ CD3+, CD8+ TILs | CRC | pCRT | Predictive of pathological downstaging | [62] | |
| FFPE tissue | ↑ Ratio of PD-1+/CD8+ TILs | Extrahepatic cholangiocarcinoma | Adjuvant CRT | ↓ OS | [63] | |
| FFPE tissue, surgical specimens | Immunoscore | CRC | pCRT | Predictive of downstaging after pCRT | [62] | |
| Surgical specimens | Immunoscore | Brain metastasis | Surgery + whole brain RT | Prognostic to OS | [62] | |
| T-cell-related | FFPE tissue | PD-L1 (+) post RT | Cervical carcinoma | Carbon-ion | ↑ PFS | [64] |
| Plasma | ↑ PD-L1 | HCC | RT | ↓ OS | [65] | |
| Plasma | T cell receptor repertoire and ↑ serum IFNβ | Lung cancer | RT + ipilimumab | Predictive of response to combination therapy | [66] | |
| Gene expression profiling | FFPE tissue | High tumour mutational burden | HNSCC | Definitive CRT | ↓ OS | [67] |
| FFPE tissue | ↑ IFN-γ signature | Bladder cancer | pCRT + surgery + CRT | ↑ Disease-specific survival | [68] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; J. Cheadle, E.; M. Illidge, T. Understanding the Effects of Radiotherapy on the Tumour Immune Microenvironment to Identify Potential Prognostic and Predictive Biomarkers of Radiotherapy Response. Cancers 2020, 12, 2835. https://doi.org/10.3390/cancers12102835
Cheng S, J. Cheadle E, M. Illidge T. Understanding the Effects of Radiotherapy on the Tumour Immune Microenvironment to Identify Potential Prognostic and Predictive Biomarkers of Radiotherapy Response. Cancers. 2020; 12(10):2835. https://doi.org/10.3390/cancers12102835
Chicago/Turabian StyleCheng, Shuhui, Eleanor J. Cheadle, and Timothy M. Illidge. 2020. "Understanding the Effects of Radiotherapy on the Tumour Immune Microenvironment to Identify Potential Prognostic and Predictive Biomarkers of Radiotherapy Response" Cancers 12, no. 10: 2835. https://doi.org/10.3390/cancers12102835
APA StyleCheng, S., J. Cheadle, E., & M. Illidge, T. (2020). Understanding the Effects of Radiotherapy on the Tumour Immune Microenvironment to Identify Potential Prognostic and Predictive Biomarkers of Radiotherapy Response. Cancers, 12(10), 2835. https://doi.org/10.3390/cancers12102835