Main Strategies for the Identification of Neoantigens


Simple Summary
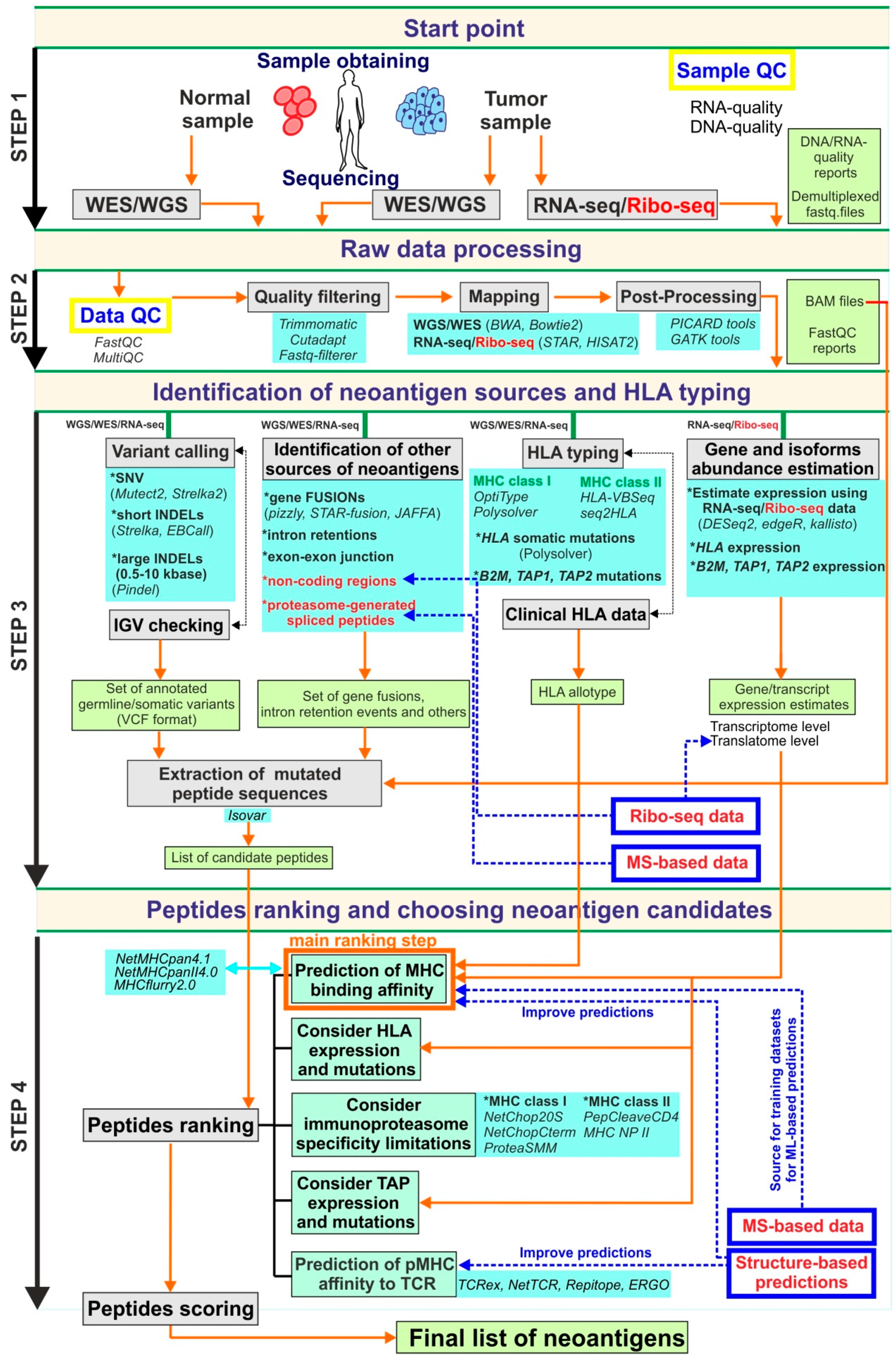
Abstract
1. Introduction
2. Genomics-Based Approaches and Current Bioinformatics Pipelines

{kind=link}
| Pipeline | Source, Required Input Data and Otput: | Workflow and Features: | Refs. |
|---|---|---|---|
| EpiToolkit 2015 | Source: http://www.epitoolkit.de (not available) Description: Web-based pipeline focused on vaccine design. It includes simplified interfaces allowing to combine tools into a workflow. Input: Not described. Output: Interactive presentation of the results as HTML and Internal representation (List of predicted peptides with scores). |
| [137] |
| FRED2 (FRamework for Epitope Detection) 2016 | Source: https://github.com/FRED-2/Fred2 Description: Computational pipeline for T-cell epitope detection and vaccine design implemented in Python. Can be extended by additional tools. Input: Sequencing reads (FASTA format). Output: Not described. |
| [138] |
| TepiTool 2016 | Source: http://tools.iedb.org/tepitool/ Description: Web-based user-friendly computational pipeline for T cell epitope prediction hosted by IEDB. It is applicable to human, chimpanzee, cow, gorilla, macaque, mouse and pig. The web-tool associated article contains a step-by-step protocol of analysis with a comprehensive description of each step, recommendations to do, and a description of anticipated results. Input: Protein sequences in single-letter amino acid code (FASTA format), the list of HLA alleles. Output: Tables with peptide sequences with predicted features. |
| [139] |
| Vaxrank 2017 | Source: https://github.com/openvax/vaxrank Description: Computational framework for selecting neoantigens for vaccine peptides based on tumor mutations, tumor RNA sequencing and HLA type data. It was designed and used in the Personalized Genomic Vaccine Phase I trial (NCT02721043). Input: Tumor mutations (VCF format), tumor RNA-seq (BAM format), patient HLA alleles. Output: Set of vaccine peptides. |
| [66,67] |
| neoantigeneR 2017 | Source: https://rdrr.io/github/tangshao2016/neoantigenR/ Description: R-based pipelines for neoantigen prediction using raw NGS data. Input: DNA-Seq, RNA-Seq, ExomeSeq (tumor and/or normal) short or long sequence reads (FASTA format), GFF annotation. Output: The list of high-affinity HLA class I binding neoantigen candidates. |
| [140] |
| CloudNeo 2017 | Source: https://github.com/TheJacksonLaboratory/CloudNeo Description: Cloud-based (implemented on CWL) workflow for neoantigen identification using NGS data. Input: VCF format (list of non-synonymous mutations), BAM format (for HLA typing). Output: HLA binding affinity predictions for all mutated peptides. |
| [141] |
| MuPexi (Mutant peptide extractor and informer) 2017 | Source: http://www.cbs.dtu.dk/services/MuPeXI/ Description: Web-based tool for neo-epitope identification using somatic mutation calls (SNV, INDELs) and obtaining information about HLA binding affinity, expression level, similarities to self-peptides and mutant allele frequency for each mutated peptide. Supplemented by brief instructions and output format description. Input: Somatic mutation calls (VCF format), list of HLA types, gene expression profile (optional). Output: Table with all tumor-specific peptides derived from substitutions, insertions and deletions with annotation (HLA binding affinity and similarity to normal peptides). |
| [142] |
| TIminer (Tumor Immunology miner) 2017 | Source: https://icbi.imed.ac.at/software/timiner/timiner.shtml (not available) Description: Computational framework that provides complex immunogenomic analysis including HLA typing, neoantigens prediction, characterization of immune infiltrates and quantification of tumor immunogenicity. Input: RNA-seq reads (FASTQ format), somatic DNA mutations (VCF format). Output: Not described. |
| [143] |
| TSNAD (Tumor-specific neoantigen detector) 2017 | Source: https://github.com/jiujiezz/tsnad Description: Pipeline with GUI allowing to identify tumor-specific mutant proteins according to GATK best practices. It provides two strategies: 1.Extraction of extracellular mutations from membrane proteins; 2. MHC affinity prediction for class I MHC. Allows us to start from raw NGS data. Input: Pair-ended sequencing data (FASTQ format) from WES. Output: List of somatic mutations with annotations, extracellular mutations of the membrane proteins and the MHC-binding information (TXT format). |
| [144] |
| INTEGRATE-neo 2017 | Source: https://github.com/ChrisMaherLab/INTEGRATE-Neo Description: The pipeline is focused on the discovery of neoantigens derived from gene fusions. Input: Reads in FASTQ format, the human reference genome in FASTA format, gene models in GenePred format, genes fusion in BEDPE format predicted by INTEGRATE. Output: BEDPE format file. |
| [88] |
| NeoepitopePred 2017 | Source: https://github.com/stjude/NeoepitopePred Description: Workflow for identification of putative neoepitopes derived from SNV and gene fusions based on WGS data. Input: FASTQ format (PE or SE) or BAM format files, Output: Not described. |
| [145] |
| Neopepsee 2018 | Source: https://sourceforge.net/projects/neopepsee/ Description: Machine learning-based neoantigen prediction tool for NGS data. Input: Raw RNA-seq data (FASTQ format) and list of somatic mutations (VCF format), clinical HLA typing (if available) Output: mutated peptide sequences and gene expression levels, determination of immunogenic neoantigens. |
| [65] |
| ScanNeo 2019 | Source: https://github.com/ylab-hi/ScanNeo Description: Computational pipeline for the identification of short and large indels-derived neoantigens utilizing RNA-seq data. ScanNeo consists of independent modules implementing three analysis steps. Input: RNA-seq data in BAM format. Output: Ranked set of neoantigens. |
| [146] |
| DeepHLApan 2019 | Source: http://biopharm.zju.edu.cn/deephlapan/ Description: Deep learning approach for neoantigen prediction considering both HLA-peptide binding (binding model) and immunogenicity (immunogenicity model) of peptide-HLA complex. Input: CSV format files with head of “Annotation,HLA,peptide”. Only HLA-A,B,C alleles. Output: Binding score (ranges from 0 to 1, the probability that peptide binds with HLA), Immunogenicity score (ranges from 0 to 1; 0.5 is the threshold to select the predicted immunogenic pHLA). |
| [147] |
| pTuneous (prioritizing tumor neoantigens from next-generation sequencing data) 2019 | Source: https://github.com/bm2-lab/pTuneos Description: In silico tool to predict the immunogenicity of SNV-derived neoepitopes that consider MHC presentation and T-cell recognition ability. It is based on experimentally validated neoantigens. It contains Pre&RecNeo module—learning-based framework allowing to predict and prioritize neoepitopes recognized by T cells and RefinedNeo module—neoepitope scoring schema allowing to evaluate the naturally processed and presented neoepitope immunogenicity Input: PairMatchDNA (WES) mode accept WES and RNA-seq sequencing data (FASTQ format), VCF mode accepts VCF format file with mutation set, expression profile (e.g., obtained by kallisto), copy number profile (e.g., obtained by sequenza). Output: TSV files (snv_neo_model.tsv and indel_neo_models.tsv) containing extracted mutated peptides derived from non-synonimous SNV and INDELs and corresponding immunity score measures. | WES mode:
| [148] |
| NeoPredPipe 2019 | Source: https://github.com/MathOnco/NeoPredPipe Description: Pipeline that provides predictions on multi-region sequence data and assessing intra-tumor heterogeneity (IHC) of the antigenic landscape of tumors. Input: Multi- or single region VCF files (with a set of somatic mutations), Patient HLA Types (optional) Output: Annotated variants, predicted neoantigens, predicted recognition potential, a summary of IHC statistics |
| [68] |
| pVACtools 2020 | Source: https://pvactools.readthedocs.io/en/latest/ Description: Computational toolkit allowing identification of altered peptides derived from SNV, INDELs, gene fusions and providing prediction of peptide-MHC binding for MHC class I and class II. Input: VCF format files, FASTA with peptides Output: A set of files containing information about predicted epitopes before and after the filtering process supplying information about binding affinity scores and other parameters. |
| [64,149] |
| ProGeo-neo 2020 | Source: https://github.com/kbvstmd/ProGeo-neo Description: Neoantigen prediction workflow that integrates genomic and mass spectrometry data. It consists of three modules: construction of customized protein sequence database, HLA allele prediction, neoantigen prediction and filtration. Input: RNA-seq data (FASTQ format), Genomic variants (VCF format), LC-MS/MS data (Raw format). Output: List of candidate peptides |
| [150] |
| Neoepiscope 2020 | Source: https://github.com/pdxgx/neoepiscope Description: Neoepitope identification pipeline that incorporates germline context and considers variant phasing for SNV and indels. Requires DNA-sequencing data. Input: Set of somatic and germline mutations (VCF format), BAM files. Output: TSV file with the information of mutations and neoepitopes |
| [80] |
| neoANT-HILL 2020 | Source: https://github.com/neoanthill/neoANT-HILL Description: User-friendly python-based toolkit that combines several pipelines that ensure fully-automated identification of potential neoantigens with a graphical interface. It allows starting from raw NGS data as well as ready-to-use variant calls. Input: Somatic variants (VCF format) and/or RNA-seq data (raw or aligned) Output: User-defined generic directory that contains variant calling data, FASTA with WT and MT sequences, predicted HLA types, gene expression estimates, tumor-infiltrating immune cells quantifications. |
| [151] |
| INeo-Epp 2020 | Source: http://www.biostatistics.online/INeo-Epp/antigen.php Description: User-friendly web-tool implementing T-cell HLA class I immunogenicity prediction method based on sequence-related amino acid features utilizing the random forest algorithm. Input: Candidate peptide sequences (8-12 aa recommended), HLA allotype Output: Table containing peptides sequences annotated with score, %rank and prediction. |
| [152] |
3. Mass Spectrometry-Based Approaches
4. Structure-Based Approaches
5. Neoantigen Peptide Databases
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chan, H.L.; Chen, P. Immune checkpoint inhibitors: Basics and challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297. [Google Scholar] [CrossRef]
- Dobry, A.S.; Zogg, C.K.; Hodi, F.S.; Smith, T.R.; Ott, P.A.; Iorgulescu, J.B. Management of metastatic melanoma: Improved survival in a national cohort following the approvals of checkpoint blockade immunotherapies and targeted therapies. Cancer Immunol. Immunother. 2018, 67, 1833–1844. [Google Scholar] [CrossRef]
- Qiu, Z.; Chen, Z.; Zhang, C.; Zhong, W. Achievements and futures of immune checkpoint inhibitors in non-small cell lung cancer. Exp. Hematol. Oncol. 2019, 8, 19. [Google Scholar] [CrossRef]
- Yan, Y.F.; Zheng, Y.F.; Ming, P.P.; Deng, X.X.; Ge, W.; Wu, Y.G. Immune checkpoint inhibitors in non-small-cell lung cancer: Current status and future directions. Brief. Funct. Genom. 2019, 18, 147–156. [Google Scholar] [CrossRef]
- Flippot, R.; Escudier, B.; Albiges, L. Immune checkpoint inhibitors: Toward new paradigms in renal cell carcinoma. Drugs 2018, 78, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Stuhler, V.; Maas, J.M.; Rausch, S.; Stenzl, A.; Bedke, J. Immune checkpoint inhibition for the treatment of renal cell carcinoma. Expert Opin. Biol. Ther. 2020, 20, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.F. Revolutionizing the landscape of colorectal cancer treatment: The Potential role of immune checkpoint inhibitors. Int. J. Cancer 2020. [Google Scholar] [CrossRef]
- Park, J.C.; Faquin, W.C.; Durbeck, J.; Faden, D.L. Immune checkpoint inhibitors in sinonasal squamous cell carcinoma. Oral Oncol. 2020, 104776. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Odunsi, K.; Coukos, G. Immune therapy opportunities in ovarian cancer. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–13. [Google Scholar] [CrossRef]
- Nakamura, Y. Biomarkers for immune checkpoint inhibitor-mediated tumor response and adverse events. Front. Med. 2019, 6. [Google Scholar] [CrossRef]
- Longo, V.; Brunetti, O.; Azzariti, A.; Galetta, D.; Nardulli, P.; Leonetti, F.; Silvestris, N. Strategies to improve cancer immune checkpoint inhibitors efficacy, other than abscopal effect: A systematic review. Cancers 2019, 11, 539. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Lyu, G.Y.; Yeh, Y.H.; Yeh, Y.C.; Wang, Y.C. Mutation load estimation model as a predictor of the response to cancer immunotherapy. NPJ Genom. Med. 2018, 3, 12. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: A systematic review and meta-analysis. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (NY) 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rao, X.; Wen, Z.; Ding, X.; Wang, X.; Xu, W.; Meng, C.; Yi, Y.; Guan, Y.; Chen, Y.; et al. Implications of driver genes associated with a high tumor mutation burden identified using next-generation sequencing on immunotherapy in hepatocellular carcinoma. Oncol. Lett. 2020, 19, 2739–2748. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Chu, Y.; Liu, Q.; Wei, J.; Liu, B. Personalized cancer neoantigen vaccines come of age. Theranostics 2018, 8, 4238–4246. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science (NY) 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, W.; Wu, J.; Lu, J.; Ding, Y.; Wu, S.; Wang, H.; Ding, D.; Mo, F.; Zhou, Z.; et al. Neoantigens derived from recurrently mutated genes as potential immunotherapy targets for gastric cancer. Biomed. Res. Int. 2019, 2019, 8103142. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Looi, K.S.; Tan, E.M. Identification of tumor-associated antigens as diagnostic and predictive biomarkers in cancer. Methods Mol. Biol. 2009, 520, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Criscitiello, C. Tumor-associated antigens in breast cancer. Breast Care (Basel) 2012, 7, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. (Hagerstown, Md. 1997) 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Sahu, A.; Singhal, U.; Chinnaiyan, A.M. Long noncoding RNAs in cancer: From function to translation. Trends Cancer 2015, 1, 93–109. [Google Scholar] [CrossRef]
- Suwinski, P.; Ong, C.; Ling, M.H.T.; Poh, Y.M.; Khan, A.M.; Ong, H.S. Advancing personalized medicine through the application of whole exome sequencing and big data analytics. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Coudray, A.; Battenhouse, A.M.; Bucher, P.; Iyer, V.R. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. Peer J. 2018, 6, e5362. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Zhao, S.; Li, C.I.; Shyr, Y.; Guo, Y. Practicability of detecting somatic point mutation from RNA high throughput sequencing data. Genomics 2016, 107, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; White, N.M.; Schmidt, H.K.; Fulton, R.S.; Tomlinson, C.; Warren, W.C.; Wilson, R.K.; Maher, C.A. Integrate: Gene fusion discovery using whole genome and transcriptome data. Genome Res. 2016, 26, 108–118. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and accurate fusion transcript detection from RNA-Seq. bioRxiv 2017. [Google Scholar] [CrossRef]
- Park, J.; Chung, Y.J. Identification of neoantigens derived from alternative splicing and RNA modification. Genom. Inform. 2019, 17, e23. [Google Scholar] [CrossRef]
- Orenbuch, R.; Filip, I.; Comito, D.; Shaman, J.; Pe’er, I.; Rabadan, R. arcasHLA: High-resolution HLA typing from RNAseq. Bioinformatics (Oxf. Engl.) 2019, 36, 33–40. [Google Scholar] [CrossRef]
- Bonsack, M.; Hoppe, S.; Winter, J.; Tichy, D.; Zeller, C.; Küpper, M.; Schitter, E.C.; Blatnik, R.; Riemer, A.B. Performance evaluation of MHC class-I binding prediction tools based on an experimentally validated MHC-peptide binding dataset. Cancer Immunol. Res. 2019. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef]
- Gfeller, D.; Bassani-Sternberg, M. Predicting antigen presentation—What could we learn from a million peptides? Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Matey-Hernandez, M.L.; Maretty, L.; Jensen, J.M.; Petersen, B.; Sibbesen, J.A.; Liu, S.; Villesen, P.; Skov, L.; Belling, K.; Have, C.T.; et al. Benchmarking the HLA typing performance of Polysolver and Optitype in 50 Danish parental trios. BMC Bioinf. 2018, 19, 239. [Google Scholar] [CrossRef] [PubMed]
- Bunce, M.; Passey, B. HLA typing by sequence-specific primers. Methods Mol. Biol. 2013, 1034, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Braunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass spectrometry profiling of HLA-Associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef]
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics 2018, 18, e1700259. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Doyle, H.A.; Mamula, M.J. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001, 22, 443–449. [Google Scholar] [CrossRef]
- Engelhard, V.H.; Altrich-Vanlith, M.; Ostankovitch, M.; Zarling, A.L. Post-translational modifications of naturally processed MHC-binding epitopes. Curr. Opin. Immunol. 2006, 18, 92–97. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.; Heck, A.J.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (NY) 2016, 354, 354–358. [Google Scholar] [CrossRef]
- Riley, T.P.; Keller, G.L.J.; Smith, A.R.; Davancaze, L.M.; Arbuiso, A.G.; Devlin, J.R.; Baker, B.M. Structure based prediction of neoantigen immunogenicity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M. Mass Spectrometry Based Immunopeptidomics for the Discovery of Cancer Neoantigens. Methods Mol. Biol. (Clifton, N.J.) 2018, 1719, 209–221. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep. 2020, 10, 3501. [Google Scholar] [CrossRef] [PubMed]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.Y.; Graubert, A.P.; et al. pVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020, 8, 409. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Rubinsteyn, A.; Hodes, I.; Kodysh, J.; Hammerbacher, J. Vaxrank: A computational tool for designing personalized cancer vaccines. bioRxiv 2017, 142919. [Google Scholar] [CrossRef]
- Rubinsteyn, A.; Kodysh, J.; Hodes, I.; Mondet, S.; Aksoy, B.A.; Finnigan, J.P.; Bhardwaj, N.; Hammerbacher, J. Computational pipeline for the PGV-001 neoantigen vaccine trial. Front. Immunol. 2018, 8. [Google Scholar] [CrossRef]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. Neopredpipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinf. 2019, 20, 264. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- David, J.K.; Maden, S.K.; Weeder, B.R.; Thompson, R.F.; Nellore, A. Putatively cancer-specific exon–exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer 2020, 2. [Google Scholar] [CrossRef]
- Smart, A.C.; Margolis, C.A.; Pimentel, H.; He, M.X.; Miao, D.; Adeegbe, D.; Fugmann, T.; Wong, K.K.; Van Allen, E.M. Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 2018, 36, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA Transcription and splicing errors as a source of cancer frameshift neoantigens for vaccines. Sci. Rep. 2019, 9, 14184. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Trans. Med. 2018, 10. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Chen, B.; Sworder, B.; Shree, T.; Liu, C.L.; Zhang, L.; Czerwinski, D.K.; Davis, M.M.; Levy, R.; et al. B-cell lymphomas present immunoglobulin neoantigens. Blood 2019, 133, 878–881. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. JNCI J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef]
- Kumai, T.; Ishibashi, K.; Oikawa, K.; Matsuda, Y.; Aoki, N.; Kimura, S.; Hayashi, S.; Kitada, M.; Harabuchi, Y.; Celis, E.; et al. Induction of tumor-reactive T helper responses by a posttranslational modified epitope from tumor protein p53. Cancer Immunol. Immunother. Cii 2014, 63, 469–478. [Google Scholar] [CrossRef]
- Wood, M.A.; Nguyen, A.; Struck, A.J.; Ellrott, K.; Nellore, A.; Thompson, R.F. Neoepiscope improves neoepitope prediction with multivariant phasing. Bioinformatics (Oxf. Engl.) 2019, 36, 713–720. [Google Scholar] [CrossRef]
- Zhao, Q.; Laverdure, J.P.; Lanoix, J.; Durette, C.; Coté, C.; Bonneil, E.; Laumont, C.M.; Gendron, P.; Vincent, K.; Courcelles, M.; et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Warden, C.D.; Adamson, A.W.; Neuhausen, S.L.; Wu, X. Detailed comparison of two popular variant calling packages for exome and targeted exon studies. PeerJ 2014, 2, e600. [Google Scholar] [CrossRef] [PubMed]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 2015, 6, 10001. [Google Scholar] [CrossRef] [PubMed]
- Callari, M.; Sammut, S.J.; De Mattos-Arruda, L.; Bruna, A.; Rueda, O.M.; Chin, S.F.; Caldas, C. Intersect-then-combine approach: Improving the performance of somatic variant calling in whole exome sequencing data using multiple aligners and callers. Genome Med. 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxf. Engl.) 2012, 28, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Sato, Y.; Chiba, K.; Okuno, Y.; Nagata, Y.; Yoshida, K.; Shiba, N.; Hayashi, Y.; Kume, H.; Homma, Y.; et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013, 41, e89. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxf. Engl.) 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-neo: A pipeline for personalized gene fusion neoantigen discovery. Bioinformatics (Oxf. Engl.) 2017, 33, 555–557. [Google Scholar] [CrossRef]
- Sijts, E.J.A.M.; Kloetzel, P.M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. CMLS 2011, 68, 1491–1502. [Google Scholar] [CrossRef]
- Rock, K.L.; Reits, E.; Neefjes, J. Present yourself! By MHC Class I and MHC Class II molecules. Trends Immunol. 2016, 37, 724–737. [Google Scholar] [CrossRef]
- Peters, B.; Bulik, S.; Tampe, R.; Van Endert, P.M.; Holzhütter, H.G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J. Immunol. (Baltimore, Md. 1950) 2003, 171, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Raghava, G.P.S. Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci. 2004, 13, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Peters, B.; Bulik, S.; Schoor, O.; Lemmel, C.; Schatz, M.M.; Kloetzel, P.M.; Rammensee, H.G.; Schild, H.; Holzhütter, H.G. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. CMLS 2005, 62, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Lata, S.; Raghava, G.P. TAPPred prediction of TAP-binding peptides in antigens. Methods Mol. Biol. 2007, 409, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Hoze, E.; Tsaban, L.; Maman, Y.; Louzoun, Y. Predictor for the effect of amino acid composition on CD4 + T cell epitopes preprocessing. J. Immunol. Methods 2013, 391, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Karosiene, E.; Dhanda, S.K.; Jurtz, V.; Edwards, L.; Nielsen, M.; Sette, A.; Peters, B. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front. Immunol. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Romero, J.M.; Jiménez, P.; Cabrera, T.; Cózar, J.M.; Pedrinaci, S.; Tallada, M.; Garrido, F.; Ruiz-Cabello, F. Coordinated downregulation of the antigen presentation machinery and HLA class I/beta2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer 2005, 113, 605–610. [Google Scholar] [CrossRef]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making sense of mass destruction: Quantitating MHC class I antigen presentation. Nat. Rev. Immunol. 2003, 3, 952–961. [Google Scholar] [CrossRef]
- Melista, E.; Rigo, K.; Pasztor, A.; Christiansen, M.; Bertinetto, F.E.; Meintjes, P.; Hague, T. Towards a new gold standard—NGS corrections to sanger SBT genotyping results. Hum. Immunol. 2015, 76, 148. [Google Scholar] [CrossRef]
- Choo, S.Y. The HLA system: Genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 2007, 48, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.C.; Zadoorian, A.; Wilson, L.O.W.; Thorne, N.P. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief. Bioinf. 2018, 19, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mai, T.H.; Nakamura, Y. Comparison of exome-based HLA class I genotyping tools: Identification of platform-specific genotyping errors. J. Hum. Genet. 2017, 62, 397–405. [Google Scholar] [CrossRef]
- Paulson, K.G.; Voillet, V.; McAfee, M.S.; Hunter, D.S.; Wagener, F.D.; Perdicchio, M.; Valente, W.J.; Koelle, S.J.; Church, C.D.; Vandeven, N.; et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun. 2018, 9, 3868. [Google Scholar] [CrossRef]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and immune escape in lung cancer evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved pan-allele prediction of MHC Class I-presented peptides by incorporating antigen processing. Cell Syst. 2020, 11, P42–P48.e7. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-source class I MHC binding affinity prediction. Cell Syst. 2018, 7, 129–132.e124. [Google Scholar] [CrossRef]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes. bioRxiv 2019, 694539. [Google Scholar] [CrossRef]
- Trolle, T.; Metushi, I.G.; Greenbaum, J.A.; Kim, Y.; Sidney, J.; Lund, O.; Sette, A.; Peters, B.; Nielsen, M. Automated benchmarking of peptide-MHC class I binding predictions. Bioinformaticsatics (Oxf. Engl.) 2015, 31, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput. Biol. 2020, 16, e1007757. [Google Scholar] [CrossRef]
- Croft, N.P.; Smith, S.A.; Pickering, J.; Sidney, J.; Peters, B.; Faridi, P.; Witney, M.J.; Sebastian, P.; Flesch, I.E.A.; Heading, S.L.; et al. Most viral peptides displayed by class I MHC on infected cells are immunogenic. Proc. Natl. Acad. Sci. USA 2019, 116, 3112–3117. [Google Scholar] [CrossRef]
- Sette, A.; Vitiello, A.; Reherman, B.; Fowler, P.; Nayersina, R.; Kast, W.M.; Melief, C.J.; Oseroff, C.; Yuan, L.; Ruppert, J.; et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J. Immunol. (Baltimore, Md. 1950) 1994, 153, 5586–5592. [Google Scholar]
- Bjerregaard, A.M.; Nielsen, M.; Jurtz, V.; Barra, C.M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. An Analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081. [Google Scholar] [CrossRef]
- Bekri, S.; Uduman, M.; Gruenstein, D.; Mei, A.H.C.; Tung, K.; Rodney-Sandy, R.; Bogen, B.; Buell, J.; Stein, R.; Doherty, K.; et al. Neoantigen synthetic peptide vaccine for multiple myeloma elicits T cell immunity in a pre-clinical model. Blood 2017, 130, 1868. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Borg, N.A.; Ely, L.K.; Beddoe, T.; Macdonald, W.A.; Reid, H.H.; Clements, C.S.; Purcell, A.W.; Kjer-Nielsen, L.; Miles, J.J.; Burrows, S.R.; et al. The CDR3 regions of an immunodominant T cell receptor dictate the ‘energetic landscape’ of peptide-MHC recognition. Nat. Immunol. 2005, 6, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.; Saulquin, X.; Reiser, J.B.; Debeaupuis, E.; Echasserieau, K.; Kissenpfennig, A.; Legoux, F.; Chouquet, A.; Le Gorrec, M.; Machillot, P.; et al. Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J. Immunol. (Baltimore, Md. 1950) 2009, 183, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yang, X.; Ko, A.; Sun, X.; Gao, M.; Zhang, Y.; Shi, A.; Mariuzza, R.A.; Weng, N.P. Sequence and structural analyses reveal distinct and highly diverse human cd8(+) tcr repertoires to immunodominant viral antigens. Cell Rep. 2017, 19, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, U.K.; Gras, S.; Kjer-Nielsen, L.; Berry, R.; Lucet, I.S.; Miles, J.J.; Tracy, S.L.; Purcell, A.W.; Bowden, D.S.; Hellard, M.; et al. An extensive antigenic footprint underpins immunodominant TCR adaptability against a hypervariable viral determinant. J. Immunol. (Baltimore, Md. 1950) 2014, 193, 5402–5413. [Google Scholar] [CrossRef]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef]
- Gielis, S.; Moris, P.; Neuter, N.D.; Bittremieux, W.; Ogunjimi, B.; Laukens, K.; Meysman, P. TCRex: A webtool for the prediction of T-cell receptor sequence epitope specificity. bioRxiv 2018, 373472. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Jessen, L.E.; Bentzen, A.K.; Jespersen, M.C.; Mahajan, S.; Vita, R.; Jensen, K.K.; Marcatili, P.; Hadrup, S.R.; Peters, B.; et al. NetTCR: Sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. bioRxiv 2018, 433706. [Google Scholar] [CrossRef]
- Ogishi, M.; Yotsuyanagi, H. Quantitative prediction of the Landscape of T cell epitope immunogenicity in sequence space. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Springer, I.; Besser, H.; Tickotsky-Moskovitz, N.; Dvorkin, S.; Louzoun, Y. Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. bioRxiv 2020, 650861. [Google Scholar] [CrossRef]
- Bi, J.; Zheng, Y.; Yan, F.; Hou, S.; Li, C. Prediction of epitope-associated TCR by using network topological similarity based on deepwalk. IEEE Access 2019, 7, 151273–151281. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, Z.; Wan, Y.; Cai, H.; Deng, L.; Li, R. The Immunogenicity and anti-tumor efficacy of a rationally designed neoantigen vaccine for B16F10 mouse melanoma. Front. Immunol. 2019, 10, 2472. [Google Scholar] [CrossRef] [PubMed]
- Ni, Q.; Zhang, F.; Liu, Y.; Wang, Z.; Yu, G.; Liang, B.; Niu, G.; Su, T.; Zhu, G.; Lu, G.; et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci. Adv. 2020, 6, eaaw6071. [Google Scholar] [CrossRef] [PubMed]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schmich, F.; Srivatsa, S.; Weidner, J.; Beerenwinkel, N.; Spang, A. Context-dependent deposition and regulation of mRNAs in P-bodies. Elife 2018, 7, e29815. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (NY) 2009, 324, 218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Fukunaga, T.; Hamada, M. Identification and analysis of ribosome-associated lncRNAs using ribosome profiling data. BMC Genom. 2018, 19, 414. [Google Scholar] [CrossRef] [PubMed]
- Schubert, B.; Brachvogel, H.P.; Jürges, C.; Kohlbacher, O. EpiToolKit—A web-based workbench for vaccine design. Bioinformatics (Oxf. Engl.) 2015, 31, 2211–2213. [Google Scholar] [CrossRef]
- Schubert, B.; Walzer, M.; Brachvogel, H.P.; Szolek, A.; Mohr, C.; Kohlbacher, O. FRED 2: An immunoinformatics framework for Python. Bioinformatics (Oxf. Engl.) 2016, 32, 2044–2046. [Google Scholar] [CrossRef]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr. Protoc. Immunol. 2016, 114, 18.19.11–18.19.24. [Google Scholar] [CrossRef]
- Tang, S.; Madhavan, S. neoantigenR: An annotation based pipeline for tumor neoantigen identification from sequencing data. bioRxiv 2017, 171843. [Google Scholar] [CrossRef]
- Bais, P.; Namburi, S.; Gatti, D.M.; Zhang, X.; Chuang, J.H. CloudNeo: A cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics (Oxf. Engl.) 2017, 33, 3110–3112. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, A.M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of neo-epitopes from tumor sequencing data. Cancer Immunol. Immunother. 2017, 66, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Tappeiner, E.; Finotello, F.; Charoentong, P.; Mayer, C.; Rieder, D.; Trajanoski, Z. TIminer: NGS data mining pipeline for cancer immunology and immunotherapy. Bioinformatics (Oxf. Engl.) 2017, 33, 3140–3141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lyu, X.; Wu, J.; Yang, X.; Wu, S.; Zhou, J.; Gu, X.; Su, Z.; Chen, S. TSNAD: An integrated software for cancer somatic mutation and tumour-specific neoantigen detection. R Soc. Open Sci. 2017, 4, 170050. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Carter, R.A.; Li, Y.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Wang, T.Y.; Wang, L.; Alam, S.K.; Hoeppner, L.H.; Yang, R. ScanNeo: Identifying indel-derived neoantigens using RNA-Seq data. Bioinformatics (Oxf. Engl.) 2019, 35, 4159–4161. [Google Scholar] [CrossRef]
- Wu, J.; Wang, W.; Zhang, J.; Zhou, B.; Zhao, W.; Su, Z.; Gu, X.; Wu, J.; Zhou, Z.; Chen, S. DeepHLApan: A deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Zhou, C.; Wei, Z.; Zhang, Z.; Zhang, B.; Zhu, C.; Chen, K.; Chuai, G.; Qu, S.; Xie, L.; Gao, Y.; et al. pTuneos: Prioritizing tumor neoantigens from next-generation sequencing data. Genome Med. 2019, 11, 67. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef]
- Li, Y.; Wang, G.; Tan, X.; Ouyang, J.; Zhang, M.; Song, X.; Liu, Q.; Leng, Q.; Chen, L.; Xie, L. ProGeo-neo: A customized proteogenomic workflow for neoantigen prediction and selection. BMC Med. Genom. 2020, 13, 52. [Google Scholar] [CrossRef]
- Coelho, A.C.M.F.; Fonseca, A.L.; Martins, D.L.; Lins, P.B.R.; da Cunha, L.M.; de Souza, S.J. neoANT-HILL: An integrated tool for identification of potential neoantigens. BMC Med. Genom. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wan, H.; Jian, X.; Li, Y.; Ouyang, J.; Tan, X.; Zhao, Y.; Lin, Y.; Xie, L. INeo-Epp: A novel T-cell HLA class-I Immunogenicity or neoantigenic epitope prediction method based on sequence-related amino acid features. Biomed. Res. Int. 2020, 2020, 5798356. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of MHC Peptides Using Mass Spectrometry For Neoantigen Discovery And Cancer Vaccine Development. Mass. Spectrum. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542. [Google Scholar] [CrossRef]
- Storkus, W.J.; Zeh, H.J., 3rd; Salter, R.D.; Lotze, M.T. Identification of T-cell epitopes: Rapid isolation of class I-presented peptides from viable cells by mild acid elution. J. Immunother. Emphas. Tumor Immunol. Off. J. Soc. Biol. Ther. 1993, 14, 94–103. [Google Scholar] [CrossRef]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-based identification of MHC-associated peptides. Cancers 2020, 12, 535. [Google Scholar] [CrossRef]
- Vigneron, N.; Stroobant, V.; Ferrari, V.; Abi Habib, J.; Van den Eynde, B.J. Production of spliced peptides by the proteasome. Mol. Immunol. 2019, 113, 93–102. [Google Scholar] [CrossRef]
- Liepe, J.; Sidney, J.; Lorenz, F.K.M.; Sette, A.; Mishto, M. Mapping the MHC class I–spliced immunopeptidome of cancer cells. Cancer Immunol. Res. 2019, 7, 62. [Google Scholar] [CrossRef]
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell Proteom. 2018, 17, 2347. [Google Scholar] [CrossRef]
- Solleder, M.; Guillaume, P.; Racle, J.; Michaux, J.; Pak, H.; Müller, M.; Coukos, G.; Bassani-Sternberg, M.; Gfeller, D. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. bioRxiv 2019, 836189. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based methods for binding mode and binding affinity prediction for peptide-MHC complexes. Curr. Top. Med. Chem. 2018, 18, 2239–2255. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Stones, D.H.; Zarling, A.L.; Willcox, C.R.; Shabanowitz, J.; Cummings, K.L.; Hunt, D.F.; Cobbold, M.; Engelhard, V.H.; Willcox, B.E. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget 2017, 8, 54160–54172. [Google Scholar] [CrossRef] [PubMed]
- Durrant, L.G.; Metheringham, R.L.; Brentville, V.A. Autophagy, citrullination and cancer. Autophagy 2016, 12, 1055–1056. [Google Scholar] [CrossRef]
- Galli-Stampino, L.; Meinjohanns, E.; Frische, K.; Meldal, M.; Jensen, T.; Werdelin, O.; Mouritsen, S. T-cell recognition of tumor-associated carbohydrates: The nature of the glycan moiety plays a decisive role in determining glycopeptide immunogenicity. Cancer Res. 1997, 57, 3214–3222. [Google Scholar]
- Schueler-Furman, O.; Altuvia, Y.; Sette, A.; Margalit, H. Structure-based prediction of binding peptides to MHC class I molecules: Application to a broad range of MHC alleles. Protein Sci. 2000, 9, 1838–1846. [Google Scholar] [CrossRef]
- Bui, H.H.; Schiewe, A.J.; von Grafenstein, H.; Haworth, I.S. Structural prediction of peptides binding to MHC class I molecules. Proteins 2006, 63, 43–52. [Google Scholar] [CrossRef]
- Yanover, C.; Bradley, P. Large-scale characterization of peptide-MHC binding landscapes with structural simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 6981. [Google Scholar] [CrossRef]
- Mukherjee, S.; Bhattacharyya, C.; Chandra, N. HLaffy: Estimating peptide affinities for Class-1 HLA molecules by learning position-specific pair potentials. Bioinformatics (Oxf. Engl.) 2016, 32, 2297–2305. [Google Scholar] [CrossRef]
- Ochoa-Garay, J.; McKinney, D.M.; Kochounian, H.H.; McMillan, M. The ability of peptides to induce cytotoxic T cells in vitro does not strongly correlate with their affinity for the H-2Ld molecule: Implications for vaccine design and immunotherapy. Mol. Immunol. 1997, 34, 273–281. [Google Scholar] [CrossRef]
- Feltkamp, M.C.; Vierboom, M.P.; Kast, W.M.; Melief, C.J. Efficient MHC class I-peptide binding is required but does not ensure MHC class I-restricted immunogenicity. Mol. Immunol. 1994, 31, 1391–1401. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Kesmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Krishna, S.; Becker, P.D.; Cocita, C.; Shu, J.; Tan, X.; Greenberg, P.D.; Klavinskis, L.S.; Blattman, J.N.; Anderson, K.S. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Nat. Acad. Sci. USA 2015, 112, E1754–E1762. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W.; Ziehm, M.; Kämper, A.; Kohlbacher, O.; Ho, S.Y. POPISK: T-cell reactivity prediction using support vector machines and string kernels. BMC Bioinf. 2011, 12, 446. [Google Scholar] [CrossRef]
- Trolle, T.; Nielsen, M. NetTepi: An integrated method for the prediction of T cell epitopes. Immunogenetics 2014, 66, 449–456. [Google Scholar] [CrossRef]
- Lanzarotti, E.; Marcatili, P.; Nielsen, M. Identification of the cognate peptide-MHC target of T cell receptors using molecular modeling and force field scoring. Mol. Immunol. 2018, 94, 91–97. [Google Scholar] [CrossRef]
- Pierce, B.G.; Weng, Z. A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 2013, 22, 35–46. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2014, 43, D405–D412. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Olsen, L.R.; Tongchusak, S.; Lin, H.; Reinherz, E.L.; Brusic, V.; Zhang, G.L. TANTIGEN: A comprehensive database of tumor T cell antigens. Cancer Immunol. Immunother. 2017, 66, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. TSNAdb: A Database for Tumor-specific neoantigens from immunogenomics data analysis. Genom. Proteom. Bioinform. 2018, 16, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.J.; Qu, Z.; Song, C.Y.; Sun, Y.; Lai, A.L.; Luo, M.Y.; Ying, Y.Z.; Meng, H.; Liang, Z.; He, Y.J.; et al. NeoPeptide: An immunoinformatic database of T-cell-defined neoantigens. Database 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, D.; Huang, P.; Jian, X.; Wan, H.; Wang, G.; Li, Y.; Ouyang, J.; Lin, Y.; Xie, L. dbPepNeo: A manually curated database for human tumor neoantigen peptides. Database 2020, 2020. [Google Scholar] [CrossRef]

| Database, Year of Appearance | Source and Description | Refs. |
|---|---|---|
| IEDB (The Immune Epitope Database) 2004 | Source: https://www.iedb.org/ Description: IEDB is one of the most powerful sources of experimental data concerning immune epitope discovery. It contains information regarding T cell epitopes of human and other organisms. It also provides tools that could be useful for neoantigen prediction. They include MHC class I and II binding predictors, including proteasome cleavage and TAP transport processing steps, as well as tools for immunogenicity predictions. For a review of this topic see [180] | [178,179,180] |
| TANTIGEN and TANTIGEN 2.0 (Tumor T-cell Antigen Database) 2009 | Source: http://projects.met-hilab.org/tadb/ Description: This database contains information about more than 1000 tumor peptides stemming from 292 different proteins. According to the description presented in [181], all peptides in the database are marked as belonging to one of four categories: (1) peptides measured in vitro to bind the HLA, but not reported to elicit either in vivo or in vitro T cell response, (2) peptides found to bind the HLA and to elicit an in vitro T cell response, (3) peptides shown to elicit in vivo tumor rejection, and (4) peptides processed and naturally presented as defined by physical detection. Moreover, peptides are annotated that are naturally processed HLA binders, e.g., peptides eluted from HLA in mass-spectrometry studies. The database also contains predicted binding peptides of 15 HLA class I and Class II. | [181] |
| TSNAdb 2018 | Source: http://biopharm.zju.edu.cn/tsnadb/ Description: TSNAdb is a freely available database developed by Wu et al. [182]. It contains results of somatic mutation identification and HLA typing analysis of 7748 tumor samples of 16 different cancer types obtained from The Cancer Genome Atlas (TCGA) and The Cancer Immunome Atlas (TCIA). Based on this data, the author predicted binding affinity between mutant/wild-type peptides and HLA class I molecules using netMHCpan v2.8/v4.0. Thus, the database contains information about 3707562/1146961 potential antigens. | [182] |
| NeoPeptide 2019 | Source: http://www.neopeptide.cn/ and https://github.com/lyotvincent/NeoPeptide Description: NeoPeptide contains information about neoantigens resulting from somatic mutations gleaned from published literature and immunological resources. As described in [183], it contains 1,818,137 epitopes obtained from more than 36,000 neoantigens that were found in different cancer types (NSCLC, breast cancer, melanoma, etc.) and specifies characteristics such as mutation site, subunit sequence, and MHC complex restriction. The database includes data concerning experimentally characterized epitopes, which are also derived from MHC binding and MHC ligand elution experiments. Information on neoantigens is cited with references to the sources. | [183] |
| dbPepNeo 2020 | Source: http://www.biostatistics.online/dbPepNeo/ Description: dbPepNeo is a manually curated database of experimentally confirmed human tumor antigens that bind specifically to HLA class I, which contains information extracted from peer-reviewed articles and the publicly available data sources. The database relies on mass spectrometry (MS) validation and specific T-cell immunoassays. The peptides were classified according to validation methods: 1. Low confidence (407794): validated by MS only; 2. Medium confidence (247): contain a somatic mutation and are validated by MS and WES/WGS; 3. High confidence (295): immunogenicity was validated directly by utilizing specific T-cell response experiments. dbPepNeo also includes the following tools: ProGeo-neo (see Table 1) and INeo-Epp, a machine learning algorithm for neoepitope immunogenicity prediction using neoantigen peptide features. | [184] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopanenko, A.V.; Kosobokova, E.N.; Kosorukov, V.S. Main Strategies for the Identification of Neoantigens. Cancers 2020, 12, 2879. https://doi.org/10.3390/cancers12102879
Gopanenko AV, Kosobokova EN, Kosorukov VS. Main Strategies for the Identification of Neoantigens. Cancers. 2020; 12(10):2879. https://doi.org/10.3390/cancers12102879
Chicago/Turabian StyleGopanenko, Alexander V., Ekaterina N. Kosobokova, and Vyacheslav S. Kosorukov. 2020. "Main Strategies for the Identification of Neoantigens" Cancers 12, no. 10: 2879. https://doi.org/10.3390/cancers12102879
APA StyleGopanenko, A. V., Kosobokova, E. N., & Kosorukov, V. S. (2020). Main Strategies for the Identification of Neoantigens. Cancers, 12(10), 2879. https://doi.org/10.3390/cancers12102879