A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway

Simple Summary
Abstract
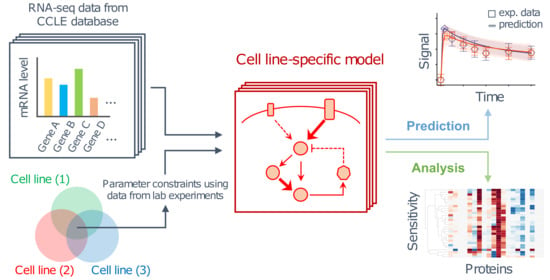
1. Introduction
2. Results
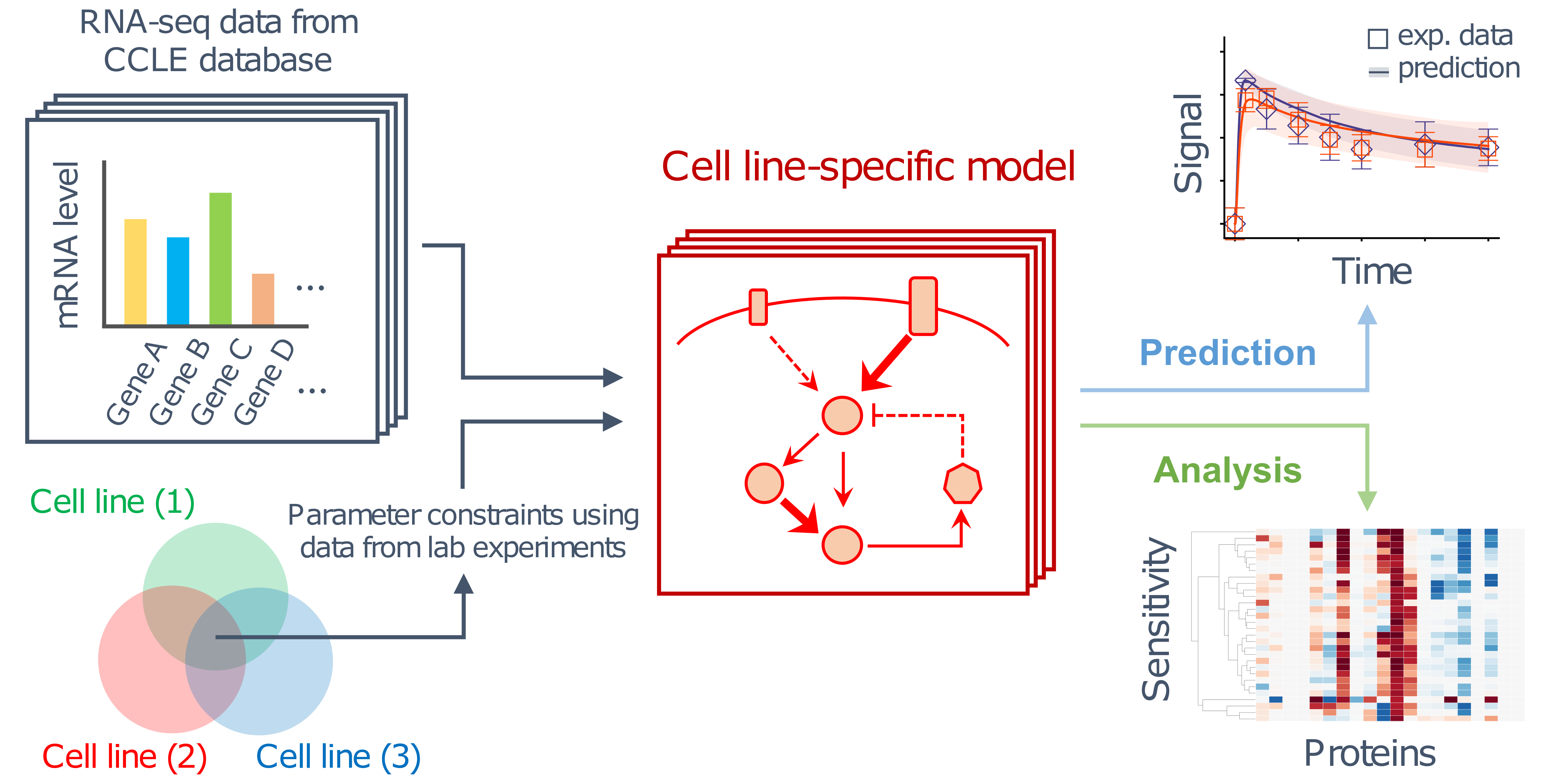
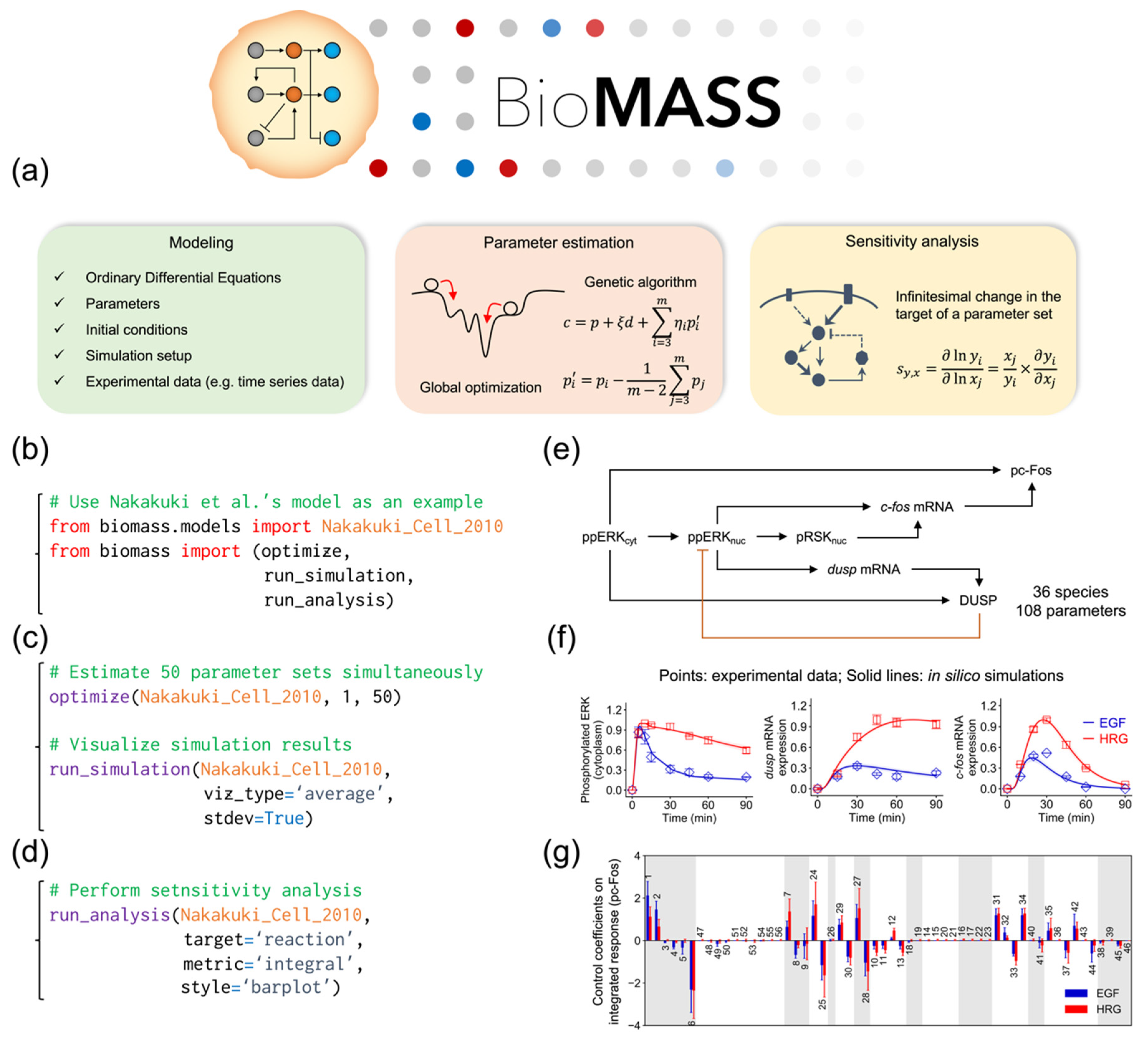
2.1. Development of BioMASS, a Framework for Modeling, Simulation, and Parameter Estimation
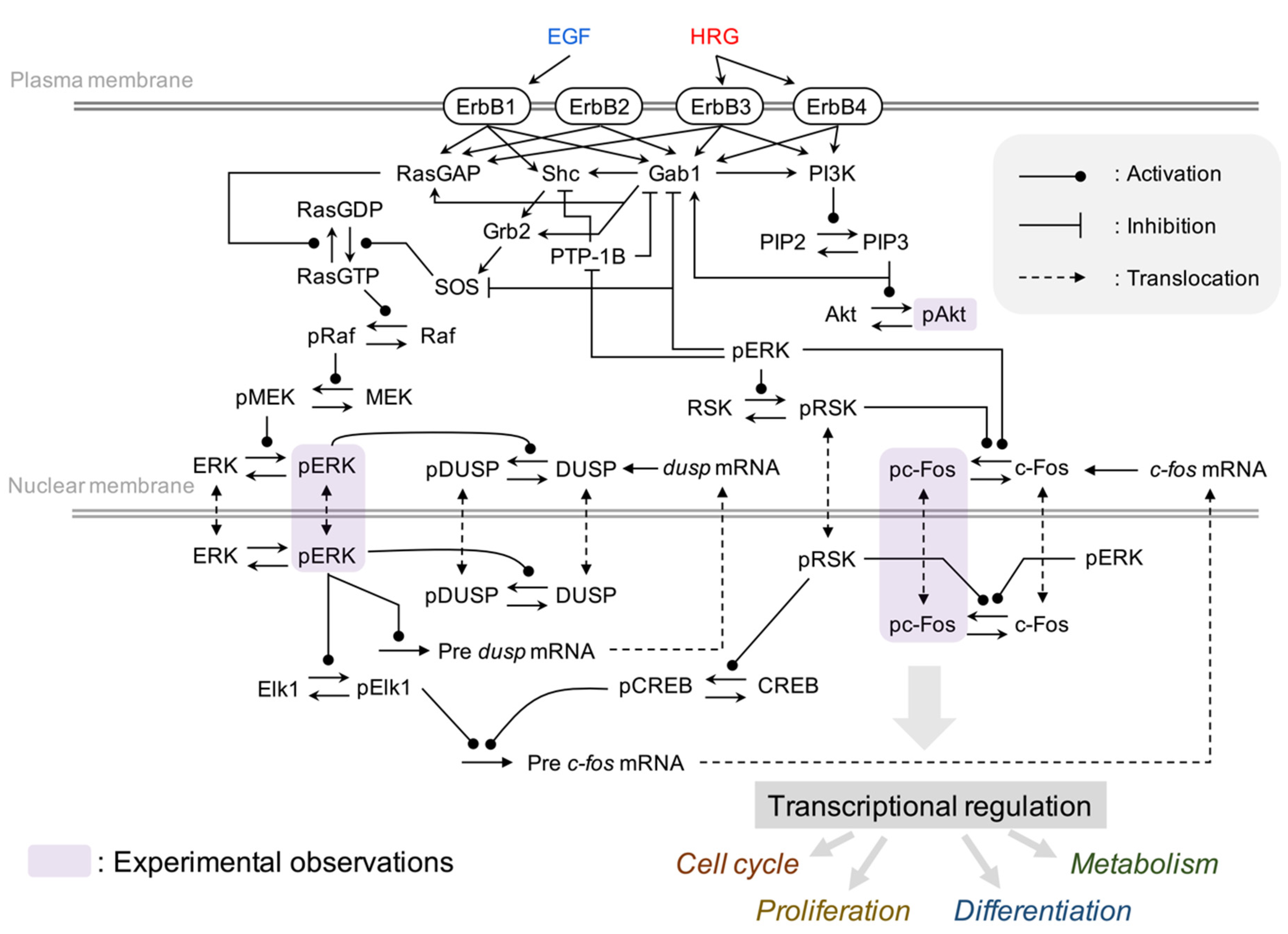
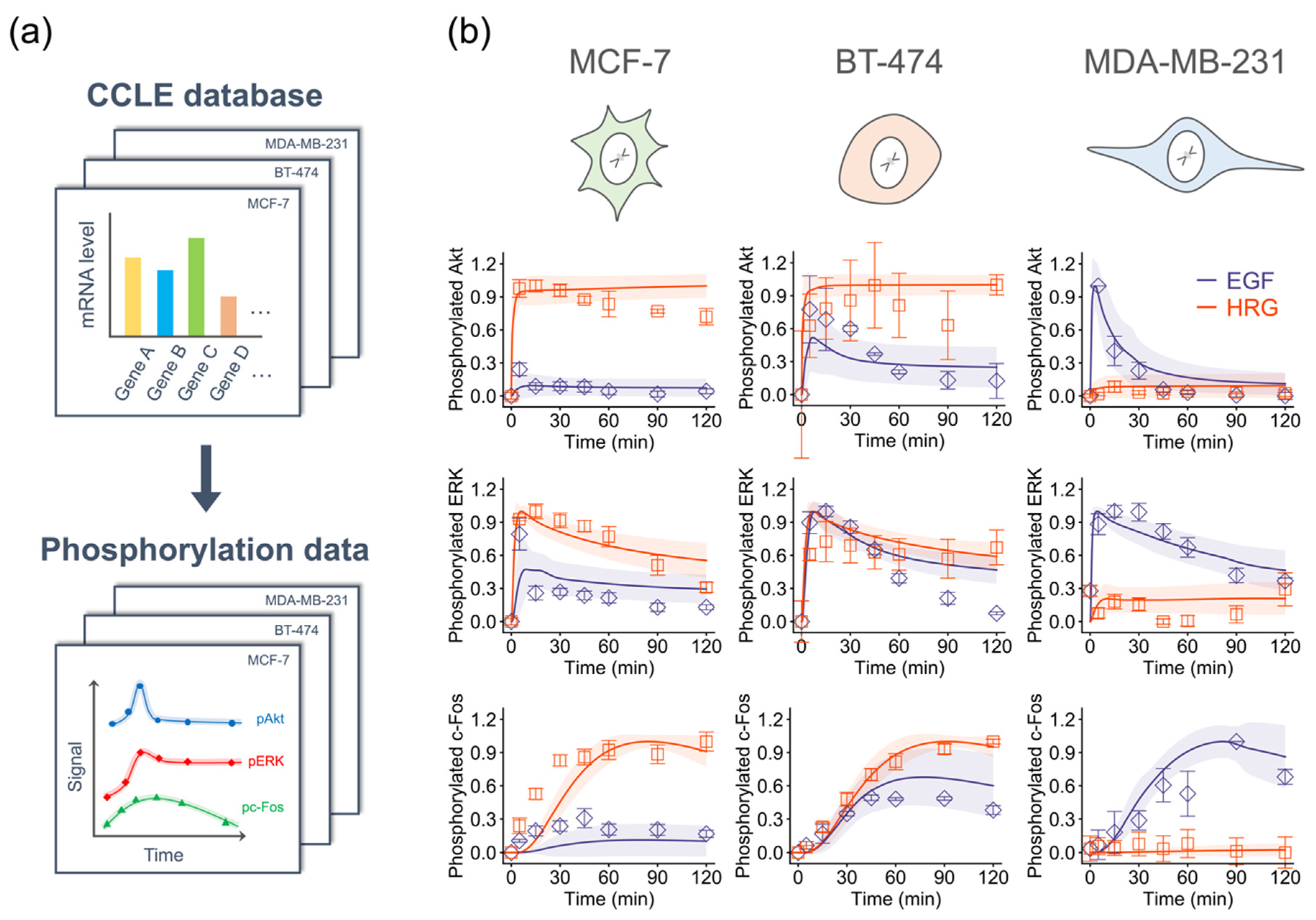
2.2. Development of Comprehensive Model of the Immediate-Early Gene Response Triggered by the ErbB Receptor in Four Breast Cancer Cell Lines
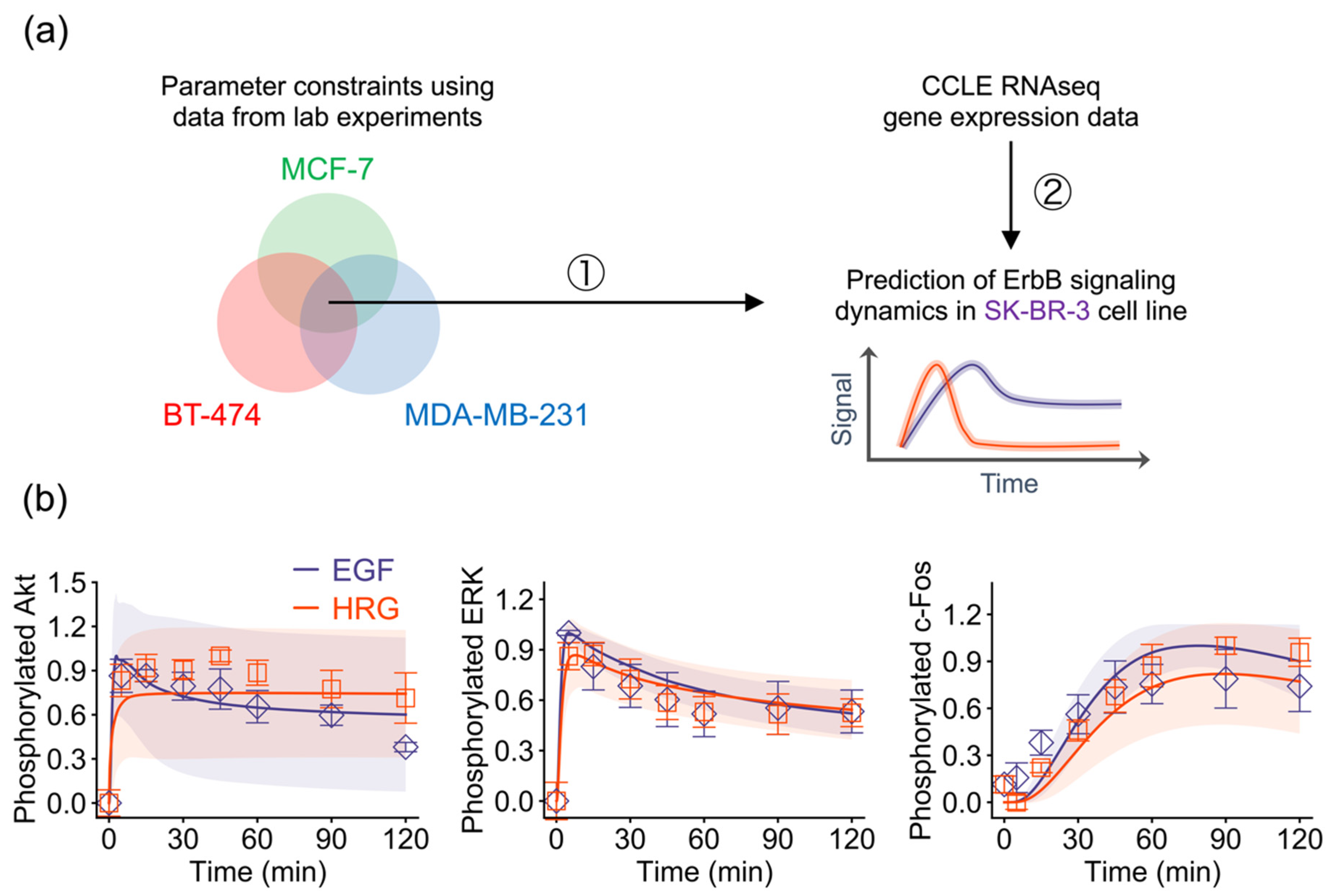
2.3. Training Model Parameters Using Gene Expression Data
2.4. Model-Based Prediction of ErbB Signaling Dynamics
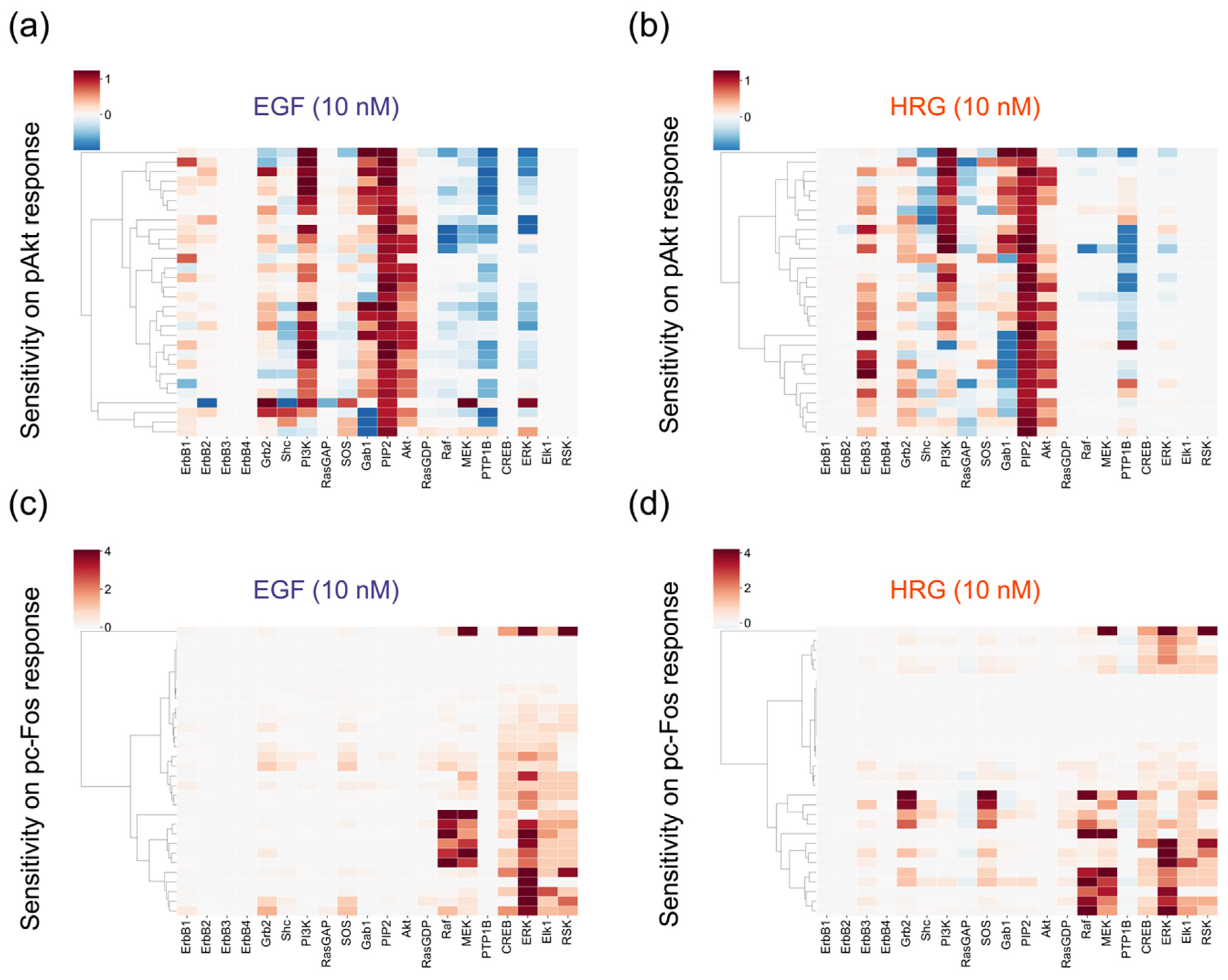
2.5. Sensitivity Analysis of Initial Values for the SK-BR-3 Cell Line
3. Discussion
4. Materials and Methods
4.1. Model Simulation and Parameter Estimation
4.2. CCLE Data
4.3. Cell Culture and Western Bloting
4.4. Data and Code Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Kudla, A.; Masson, K.; Kalra, A.; Curley, M.; Finn, G.; Pace, E.; Harms, B.; Kim, J.; Kearns, J.; et al. Systems biology driving drug development: From design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). NPJ Syst. Biol. Appl. 2017, 3, 16034. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.A.; Fisher, J. Executable cancer models: Successes and challenges. Nat. Rev. Cancer 2020, 20, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Alkan, O.; Schoeberl, B.; Shah, M.; Koshkaryev, A.; Heinemann, T.; Drummond, D.C.; Yaffe, M.B.; Raue, A. Modeling chemotherapy-induced stress to identify rational combination therapies in the DNA damage response pathway. Sci. Signal. 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Tyson, J.J.; Novak, B. A Dynamical Paradigm for Molecular Cell Biology. Trends Cell Biol. 2020, 1–12. [Google Scholar] [CrossRef]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Schoeberl, B.; Pace, E.A.; Fitzgerald, J.B.; Harms, B.D.; Xu, L.; Nie, L.; Linggi, B.; Kalra, A.; Paragas, V.; Bukhalid, R.; et al. Therapeutically targeting ErbB3: A key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef]
- Chen, W.W.; Schoeberl, B.; Jasper, P.J.; Niepel, M.; Nielsen, U.B.; Lauffenburger, D.A.; Sorger, P.K. Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol. Syst. Biol. 2009, 5. [Google Scholar] [CrossRef]
- Hass, H.; Masson, K.; Wohlgemuth, S.; Paragas, V.; Allen, J.E.; Sevecka, M.; Pace, E.; Timmer, J.; Stelling, J.; MacBeath, G.; et al. Predicting ligand-dependent tumors from multi-dimensional signaling features. NPJ Syst. Biol. Appl. 2017, 3, 27. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Shimodaira, H.; Ide, K.; Nakakuki, T.; Tani, Y.; Takahashi, K.; Yumoto, N.; Hatakeyama, M. Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J. Biol. Chem. 2007, 282, 4045–4056. [Google Scholar] [CrossRef] [PubMed]
- Nakakuki, T.; Birtwistle, M.R.; Saeki, Y.; Yumoto, N.; Ide, K.; Nagashima, T.; Brusch, L.; Ogunnaike, B.A.; Okada-Hatakeyama, M.; Kholodenko, B.N. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 2010, 141, 884–896. [Google Scholar] [CrossRef]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular, interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Ryl, T.; Kuchen, E.E.; Bell, E.; Shao, C.; Flórez, A.F.; Mönke, G.; Gogolin, S.; Friedrich, M.; Lamprecht, F.; Westermann, F.; et al. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 2017, 5, 237–250.e8. [Google Scholar] [CrossRef]
- Imoto, H.; Okada, M. Signal-dependent regulation of early-response genes and cell cycle: A quantitative view. Curr. Opin. Syst. Biol. 2019, 15, 100–108. [Google Scholar] [CrossRef]
- Mina, M.; Magi, S.; Jurman, G.; Itoh, M.; Kawaji, H.; Lassmann, T.; Arner, E.; Forrest, A.R.R.; Carninci, P.; Hayashizaki, Y.; et al. Promoter-level expression clustering identifies time development of transcriptional regulatory cascades initiated by ERBB receptors in breast cancer cells. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017. [Google Scholar] [CrossRef]
- Birtwistle, M.R.; Hatakeyama, M.; Yumoto, N.; Ogunnaike, B.A.; Hoek, J.B.; Kholodenko, B.N. Ligand-dependent responses of the ErbB signaling network: Experimental and modeling analyses. Mol. Syst. Biol. 2007, 3. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Behar, M.; Inoue, K.; Hiroshima, M.; Yasuda, T.; Nagashima, T.; Kimura, S.; Sanjo, H.; Maeda, S.; Yumoto, N.; et al. Positive feedback within a kinase signaling complex functions as a switch mechanism for NF-κB activation. Science 2014, 344, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.Y.-C.; Choi, Y.S.; Ma, W.; Pomerening, J.R.; Tang, C.; Ferrell, J.E. Robust, Tunable Biological Oscillations from Interlinked Positive and Negative Feedback Loops. Science 2008, 321, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Iwamoto, K.; Yumoto, N.; Hiroshima, M.; Nagashima, T.; Ohki, R.; Garcia-Munoz, A.; Volinsky, N.; Von Kriegsheim, A.; Sako, Y.; et al. Transcriptionally inducible pleckstrin homology-like domain, family a, member 1, attenuates ERBB receptor activity by inhibiting receptor oligomerization. J. Biol. Chem. 2018, 293, 2206–2218. [Google Scholar] [CrossRef]
- Raue, A.; Schilling, M.; Bachmann, J.; Matteson, A.; Schelke, M.; Kaschek, D.; Hug, S.; Kreutz, C.; Harms, B.D.; Theis, F.J.; et al. Lessons Learned from Quantitative Dynamical Modeling in Systems Biology. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Heinrich, R.; Neel, B.G.; Rapoport, T.A. Mathematical models of protein kinase signal transduction. Mol. Cell 2002, 9, 957–970. [Google Scholar] [CrossRef]
- Kholodenko, B.; Yaffe, M.B.; Kolch, W. Computational approaches for analyzing information flow in biological networks. Sci. Signal. 2012, 8. [Google Scholar] [CrossRef]
- Raue, A.; Steiert, B.; Schelker, M.; Kreutz, C.; Maiwald, T.; Hass, H.; Vanlier, J.; Tönsing, C.; Adlung, L.; Engesser, R.; et al. Data2Dynamics: A modeling environment tailored to parameter estimation in dynamical systems. Bioinformatics 2015, 31, 3558–3560. [Google Scholar] [CrossRef]
- Ono, I.; Satoh, H.; Kobayashi, S. A robust real-coded genetic algorithm using unimodal normal distribution crossover augmented by uniform crossover: Effects of self-adaptation of crossover probabilities. In Proceedings of the 1st Annual Conference on Genetic and Evolutionary Computation; Terence, S., Jason, H.M., Eds.; Association for Computing Machinery: New York, NY, USA, 1999; Volume 1, pp. 496–503. [Google Scholar]
- Sato, H.; Ono, I.; Kobayashi, S. A New Generation Alternation Model of Genetic Algorithms and Its Assessment. J. Jpn. Soc. Artif. Intell. 1997, 12, 734–744. [Google Scholar]
- Kimura, S.; Ono, I.; Kita, H.; Kobayashi, S. An extension of UNDX based on guidelines for designing crossover operators: Proposition and evaluation of ENDX. Trans. Soc. Instrum. Control Eng. 2000, 36, 1162–1171. [Google Scholar] [CrossRef][Green Version]
- Kimura, S.; Konagaya, A. A Genetic Algorithm with Distance Independent Diversity Control for High Dimensional Function Optimization. J. Jpn. Soc. Artif. Intell. 2003, 18, 193–202. [Google Scholar]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, F.; Kessler, T.; Weindl, D.; Shadrin, A.; Schmiester, L.; Hache, H.; Muradyan, A.; Schütte, M.; Lim, J.H.; Heinig, M.; et al. Efficient Parameter Estimation Enables the Prediction of Drug Response Using a Mechanistic Pan-Cancer Pathway Model. Cell Syst. 2018, 7, 567–579.e6. [Google Scholar] [CrossRef] [PubMed]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Phil. Trans. R. Soc. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [PubMed]
- Zi, Z. Sensitivity analysis approaches applied to systems biology models. IET Syst. Biol. 2011, 5, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar] [CrossRef]
- Giudice, G.; Petsalaki, E. Proteomics and phosphoproteomics in precision medicine: Applications and challenges. Brief. Bioinform. 2017, 20, 767–777. [Google Scholar] [CrossRef]
- Hidalgo, M.R.; Cubuk, C.; Amadoz, A.; Salavert, F.; Carbonell-Caballero, J.; Dopazo, J. High throughput estimation of functional cell activities reveals disease mechanisms and predicts relevant clinical outcomes. Oncotarget 2017, 8, 5160–5178. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, C.; Hidalgo, M.R.; Amadoz, A.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Gene expression integration into pathway modules reveals a pan-cancer metabolic landscape. Cancer Res. 2018, 78, 6059–6072. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.E.; Peña, J.M.; Jayamohan, J.; Jérusalem, A. Mechanistic models versus machine learning, a fight worth fighting for the biological community? Biol. Lett. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.M.; Verveer, P.J.; Bastiaens, P.I.H. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 2007, 9, 324–330. [Google Scholar] [CrossRef]
- Purvis, J.E.; Lahav, G. Encoding and decoding cellular information through signaling dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef]
- Schnitt, S.J. Classification and prognosis of invasive breast cancer: From morphology to molecular taxonomy. Mod. Pathol. 2010, 23, 60–64. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Saez-Rodriguez, J.; Blüthgen, N. Personalized signaling models for personalized treatments. Mol. Syst. Biol. 2020, 16. [Google Scholar] [CrossRef]
- Hindmarsh, A.C.; Brown, P.N.; Grant, K.E.; Lee, S.L.; Serban, R.; Shumaker, D.E.; Woodward, C.S. SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 2005, 31, 363–396. [Google Scholar] [CrossRef]
- Rackauckas, C.; Nie, Q. DifferentialEquations.jl—A Performant and Feature-Rich Ecosystem for Solving Differential Equations in Julia. J. Open Res. Softw. 2017, 5. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kiyatkin, A.; Aksamitiene, E. Multistrip western blotting to increase quantitative data output. Methods Mol. Biol. 2009, 536, 149–161. [Google Scholar] [CrossRef]
- Degasperi, A.; Birtwistle, M.R.; Volinsky, N.; Rauch, J.; Kolch, W.; Kholodenko, B.N. Evaluating strategies to normalise biological replicates of western blot data. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Content |
|---|---|
| name2idx/ | Names of model parameters and species |
| set_model.py | Differential equations, parameters and initial conditions |
| observable.py | Observables, simulations and experimental data |
| viz.py | Plotting options for customizing figure properties |
| set_serach_param.py | Model parameters to optimize and search region |
| fitness.py | An objective function to be minimized |
| reaction_network.py | Reaction indices grouped according to biological processes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imoto, H.; Zhang, S.; Okada, M. A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway. Cancers 2020, 12, 2878. https://doi.org/10.3390/cancers12102878
Imoto H, Zhang S, Okada M. A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway. Cancers. 2020; 12(10):2878. https://doi.org/10.3390/cancers12102878
Chicago/Turabian StyleImoto, Hiroaki, Suxiang Zhang, and Mariko Okada. 2020. "A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway" Cancers 12, no. 10: 2878. https://doi.org/10.3390/cancers12102878
APA StyleImoto, H., Zhang, S., & Okada, M. (2020). A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway. Cancers, 12(10), 2878. https://doi.org/10.3390/cancers12102878