Cancer Predisposition Genes in Cancer-Free Families


 , ,
, , 

Simple Summary
Abstract
1. Introduction
2. Results
2.1. Low-Risk Variants
2.2. Suggested Cancer Predisposition Genes
2.3. High-Risk Breast, Colorectal, and Prostate Cancer Predisposition Genes
3. Discussion
4. Materials and Methods
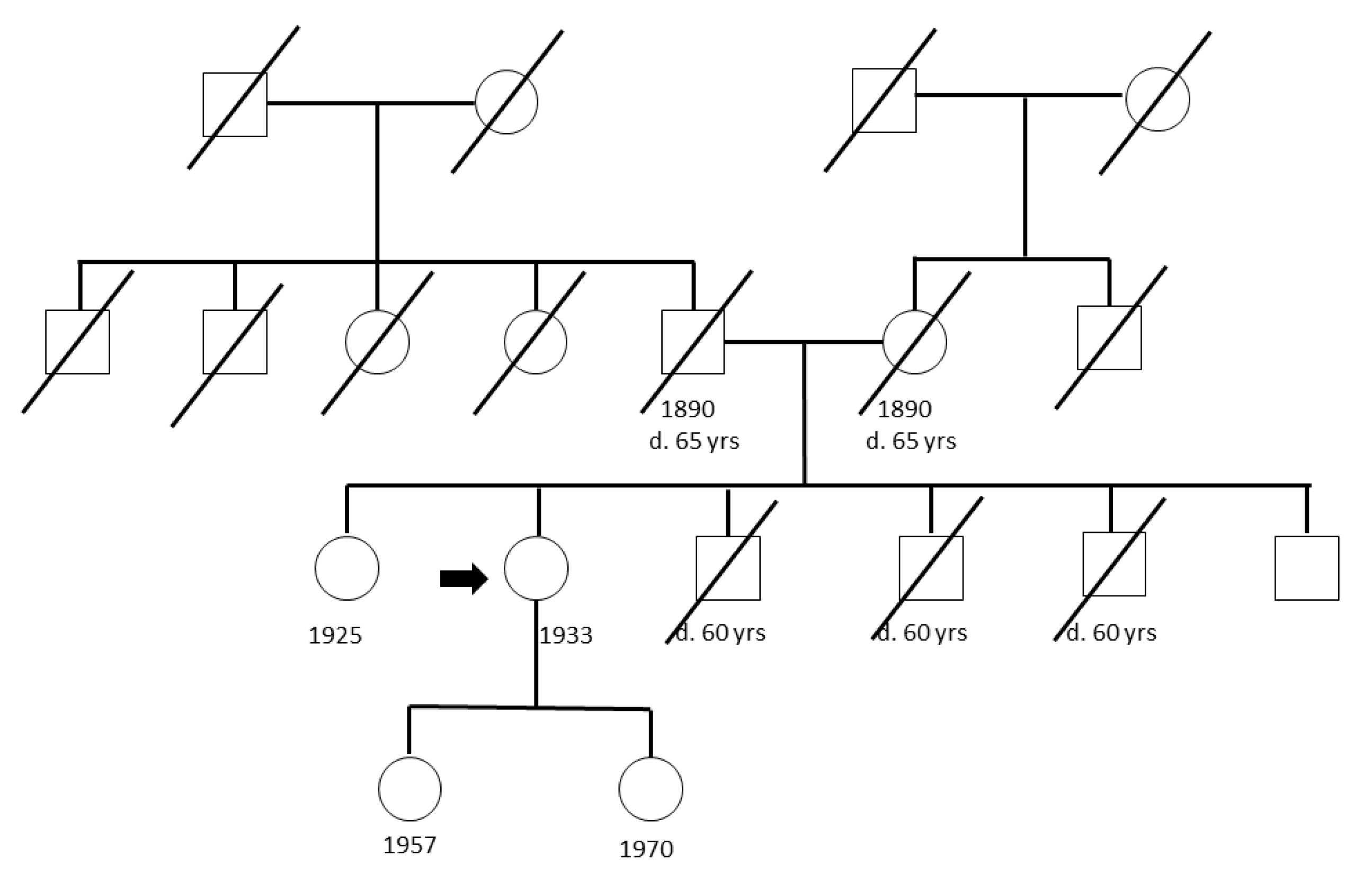
4.1. Study Populations
4.2. Ethics Statement
4.3. Whole-Genome Sequencing
4.4. Low-Risk Variants
4.5. Suggested Cancer Predisposition Genes
4.6. Variants in High-Risk Genes of Breast, Colorectal, and Prostate Cancer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frank, C.; Sundquist, J.; Yu, H.; Hemminki, A.; Hemminki, K. Concordant and discordant familial cancer: Familial risks, proportions and population impact. Int. J. Cancer 2017, 140, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from sweden, denmark, and finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Moller, S.; Unger, R.H.; et al. Familial risk and heritability of cancer among twins in nordic countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Artomov, M.; Joseph, V.; Tiao, G.; Thomas, T.; Schrader, K.; Klein, R.J.; Kiezun, A.; Gupta, N.; Margolin, L.; Stratigos, A.J.; et al. Case-control analysis identifies shared properties of rare germline variation in cancer predisposing genes. Eur. J. Hum. Genet. 2019, 27, 824–828. [Google Scholar] [CrossRef]
- Sampson, J.N.; Wheeler, W.A.; Yeager, M.; Panagiotou, O.; Wang, Z.; Berndt, S.I.; Lan, Q.; Abnet, C.C.; Amundadottir, L.T.; Figueroa, J.D.; et al. Analysis of heritability and shared heritability based on genome-wide association studies for thirteen cancer types. J. Natl. Cancer Inst. 2015, 107, e279. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R.S. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (crc) is achievable for a high proportion of familial crc by exome sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef]
- Palomaki, G.E. Is it time for brca1/2 mutation screening in the general adult population?: Impact of population characteristics. Genet. Med. 2015, 17, 24–26. [Google Scholar] [CrossRef]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018, 173, 355–370. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Schmit, S.L.; Edlund, C.K.; Schumacher, F.R.; Gong, J.; Harrison, T.A.; Huyghe, J.R.; Qu, C.; Melas, M.; Van Den Berg, D.J.; Wang, H.; et al. Novel common genetic susceptibility loci for colorectal cancer. J. Natl. Cancer Inst. 2019, 111, 146–157. [Google Scholar] [CrossRef]
- Yu, H.; Frank, C.; Sundquist, J.; Hemminki, A.; Hemminki, K. Common cancers share familial susceptibility: Implications for cancer genetics and counselling. J. Med. Genet. 2017, 54, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Pinese, M.; Lacaze, P.; Rath, E.M.; Stone, A.; Brion, M.J.; Ameur, A.; Nagpal, S.; Puttick, C.; Husson, S.; Degrave, D.; et al. The medical genome reference bank contains whole genome and phenotype data of 2570 healthy elderly. Nat. Commun. 2020, 11, e435. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lilyquist, J.; Ruddy, K.J.; Vachon, C.M.; Couch, F.J. Common genetic variation and breast cancer risk-past, present, and future. Cancer. Epidemiol. Biomark. Prev. 2018, 27, 380–394. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef]
- Wei, R.; Yao, Y.; Yang, W.; Zheng, C.H.; Zhao, M.; Xia, J. Dbcpg: A web resource for cancer predisposition genes. Oncotarget 2016, 7, 37803–37811. [Google Scholar] [CrossRef]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Forsti, A. Familial cancer variant prioritization pipeline version 2 (fcvppv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef]
- Czene, K.; Lichtenstein, P.; Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the swedish family-cancer database. Int. J. Cancer 2002, 99, 260–266. [Google Scholar] [CrossRef]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Forsti, A.; Ali, M.; van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7, 12050. [Google Scholar] [CrossRef] [PubMed]
- Cremers, R.G.; Galesloot, T.E.; Aben, K.K.; van Oort, I.M.; Vasen, H.F.; Vermeulen, S.H.; Kiemeney, L.A. Known susceptibility snps for sporadic prostate cancer show a similar association with "hereditary" prostate cancer. Prostate 2015, 75, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Archambault, A.N.; Su, Y.R.; Jeon, J.; Thomas, M.; Lin, Y.; Conti, D.V.; Win, A.K.; Sakoda, L.C.; Lansdorp-Vogelaar, I.; Peterse, E.F.; et al. Cumulative burden of colorectal cancer-associated genetic variants is more strongly associated with early-onset vs late-onset cancer. Gastroenterology 2019, 158, 1274–1286. [Google Scholar] [CrossRef] [PubMed]
- Cust, A.E.; Drummond, M.; Kanetsky, P.A.; Mann, G.J.; Schmid, H.; Hopper, J.L.; Aitken, J.F.; Armstrong, B.K.; Giles, G.G.; Holland, E.; et al. Assessing the incremental contribution of common genomic variants to melanoma risk prediction in two population-based studies. J. Invest. Derm. 2018, 138, 2617–2624. [Google Scholar] [CrossRef]
- Weigl, K.; Thomsen, H.; Balavarca, Y.; Hellwege, J.N.; Shrubsole, M.J.; Brenner, H. Genetic risk score is associated with prevalence of advanced neoplasms in a colorectal cancer screening population. Gastroenterology 2018, 155, 88–98. [Google Scholar] [CrossRef]
- Ji, J.; Sundquist, K.; Sundquist, J.; Hemminki, K. Comparability of cancer identification among death registry, cancer registry and hospital discharge registry. Int. J. Cancer 2012, 131, 2085–2093. [Google Scholar] [CrossRef]
- Andersen, M.M.; Eriksen, P.S.; Morling, N. Cluster analysis of european y-chromosomal str haplotypes using the discrete laplace method. Forensic Sci. Int. Genet. 2014, 11, 182–194. [Google Scholar] [CrossRef]
- Heath, S.C.; Gut, I.G.; Brennan, P.; McKay, J.D.; Bencko, V.; Fabianova, E.; Foretova, L.; Georges, M.; Janout, V.; Kabesch, M.; et al. Investigation of the fine structure of european populations with applications to disease association studies. Eur. J. Hum. Genet. 2008, 16, 1413–1429. [Google Scholar] [CrossRef]
- Mielnik-Sikorska, M.; Daca, P.; Malyarchuk, B.; Derenko, M.; Skonieczna, K.; Perkova, M.; Dobosz, T.; Grzybowski, T. The history of slavs inferred from complete mitochondrial genome sequences. PLoS ONE 2013, 8, e54360. [Google Scholar] [CrossRef]
- Nelis, M.; Esko, T.; Magi, R.; Zimprich, F.; Zimprich, A.; Toncheva, D.; Karachanak, S.; Piskackova, T.; Balascak, I.; Peltonen, L.; et al. Genetic structure of europeans: A view from the north-east. PLoS ONE 2009, 4, e5472. [Google Scholar] [CrossRef]
- Guo, M.H.; Plummer, L.; Chan, Y.M.; Hirschhorn, J.N.; Lippincott, M.F. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am. J. Hum. Genet. 2018, 103, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, C.; Sud, A.; Houlston, R.S. Cancer genetics, precision prevention and a call to action. Nat. Genet. 2018, 50, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acids. Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. Dbsnp: A database of single nucleotide polymorphisms. Nucleic. Acids. Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. Dbnsfp v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site snvs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013, 7, e20. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic. Acids. Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Castera, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 hboc families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef]
- Haukoos, J.S.; Lewis, R.J. Advanced statistics: Bootstrapping confidence intervals for statistics with "difficult" distributions. Acad. Emerg. Med. 2005, 12, 360–365. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cancer | SNPID | Gene | Risk Allele | Frequency | OR | 95% CI | p1 | ||
|---|---|---|---|---|---|---|---|---|---|
| GnomAD | CFF | ||||||||
| BC | rs10474352 | ARRDC3 | C | 0.83 | 0.74 | 0.56 | 0.36 | 0.87 | 0.0097 |
| rs16886181 | MAP3K1 | C | 0.17 | 0.08 | 0.41 | 0.20 | 0.85 | 0.0165 | |
| rs206966 | RP1-166H1.2 | T | 0.17 | 0.25 | 1.67 | 1.07 | 2.61 | 0.0248 | |
| rs2992756 | KLHDC7A | T | 0.49 | 0.37 | 0.62 | 0.41 | 0.93 | 0.0197 | |
| rs653465 | SLC4A7 | C | 0.47 | 0.57 | 1.48 | 1.00 | 2.20 | 0.0489 | |
| rs7072776 | DNAJC1 | A | 0.28 | 0.19 | 0.58 | 0.35 | 0.96 | 0.0348 | |
| rs889312 | MAP3K1 | C | 0.29 | 0.18 | 0.53 | 0.32 | 0.89 | 0.0154 | |
| CRC | rs17816465 | GREM1 | A | 0.20 | 0.10 | 0.43 | 0.23 | 0.84 | 0.0125 |
| PC | rs10460109 | TSHZ1 | T | 0.42 | 0.56 | 1.72 | 1.16 | 2.55 | 0.0067 |
| rs3850699 | TRIM8 | A | 0.68 | 0.56 | 0.6 | 0.41 | 0.89 | 0.0110 | |
| rs28607662 | TCF4 | C | 0.09 | 0.17 | 1.96 | 1.16 | 3.31 | 0.0118 | |
| rs2066827 | CDKN1B | T | 0.75 | 0.86 | 2.05 | 1.17 | 3.61 | 0.0125 | |
| rs2680708 | RNF43 | G | 0.60 | 0.48 | 0.62 | 0.42 | 0.91 | 0.0153 | |
| rs33984059 | RFX7 | A | 0.98 | 0.94 | 0.37 | 0.16 | 0.85 | 0.0193 | |
| rs12155172 | LINC01162 | A | 0.24 | 0.33 | 1.62 | 1.07 | 2.45 | 0.0216 | |
| rs6465657 | LMTK2 | C | 0.46 | 0.57 | 1.56 | 1.05 | 2.31 | 0.0265 | |
| rs12543663 | PCAT1 | C | 0.31 | 0.41 | 1.56 | 1.05 | 2.32 | 0.0270 | |
| rs9364554 | SLC22A3 | T | 0.27 | 0.19 | 0.60 | 0.37 | 1.00 | 0.0478 | |
| Cancer | No. Risk Alleles | 1000 Genomes No. | CFF No. | OR | 95%CI | p | |
|---|---|---|---|---|---|---|---|
| BC | ≤87 | 73 | 19 | 1.00 | - | - | |
| 88–91 | 72 | 12 | 0.64 | 0.29 | 1.42 | 0.27 | |
| 92–96 | 77 | 11 | 0.55 | 0.24 | 1.23 | 0.15 | |
| >96 | 72 | 9 | 0.48 | 0.20 | 1.13 | 0.09 | |
| p-trend = 0.07 | |||||||
| CRC | ≤71 | 75 | 13 | 1.00 | |||
| 72–76 | 90 | 13 | 0.83 | 0.36 | 1.91 | 0.67 | |
| 77–80 | 69 | 16 | 1.34 | 0.60 | 2.98 | 0.48 | |
| >80 | 60 | 9 | 0.87 | 0.35 | 2.16 | 0.76 | |
| p-trend = 0.88 | |||||||
| PC | ≤89 | 91 | 10 | 1.00 | |||
| 90–93 | 64 | 10 | 1.42 | 0.56 | 3.62 | 0.46 | |
| 94–97 | 86 | 10 | 1.06 | 0.42 | 2.67 | 0.90 | |
| >97 | 53 | 21 | 3.61 | 1.58 | 8.23 | 0.0023 | |
| p-trend = 0.0055 | |||||||
| Source of CPGs | CFF No. Variants | P CFF | ExAC No. Variants | P ExAC | OR | 95%CI | |
|---|---|---|---|---|---|---|---|
| Wei [18] non-synonymous | 54 | 67 % | 23419 | 63 % | 1.21 | 0.77 | 1.91 |
| Wei [18] LoF | 2 | 6 % | 3675 | 15 % | 0.35 | 0.00 | 0.53 |
| Rahman [17] non-synonymous | 18 | 31 % | 5619 | 22 % | 1.58 | 0.87 | 2.83 |
| Rahman [17] LoF | 0 | 0 % | 791 | 4 % | |||
| Gene | Missense + LoF Variants in ExAC | Missense + LoF Variants in CFF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total No. | No. Pathogenic | p ExAC 1 | SNP ID | Chr | Position | Ref/Alt | Prevalence ExAC NFE | CADD | Positive Conservation Scores | Positive Prediction Tools | ClinVar Significance | |
| BRCA2 | 691 | 40 | 0.112% | rs397507270 | 13 | 32907128 | A/G | 1.51 × 10−5 | 0.11 | 0 | 1 | Likely benign/US |
| rs56087561 | 13 | 32913562 | A/C | 3.65 × 10−4 | 24.1 | 2 | 5 | Benign | ||||
| rs80358768 | 13 | 32913947 | C/T | 3.45 × 10−4 | 0.2 | 0 | 1 | Benign | ||||
| APC | 481 | 2 | 0.003% | rs748940586 | 5 | 112178309 | A/C | 1.51 × 10−5 | 22.7 | 3 | 8 | US |
| No dbSNP | 5 | 112178460 | GTAT/G | . | 21.8 | . | . | - | ||||
| MLH1 | 167 | 3 | 0.008% | rs41294980 | 3 | 37067306 | G/A | 1.18 × 10−3 | 7.3 | 1 | 0/4 2 | Benign |
| rs63751225 | 3 | 37090075 | T/C | 1.80 × 10−4 | 22.1 | 3 | 4 | US | ||||
| MSH2 | 246 | 4 | 0.006% | rs116117580 | 2 | 47739533 | G/A | 1.99 × 10−2 | 0.003 | 0 | 1 | Not provided |
| MSH6 | 359 | 8 | 0.017% | rs752887988 | 2 | 48010377 | C/T | 0 | 33 | 3 | 7 | - |
| rs267608075 | 2 | 48028282 | A/T | 1.83 × 10−4 | 13.0 | 3 | 5 | Benign/US | ||||
| MUTYH | 174 | 12 | 0.079% | rs36053993 | 1 | 45797228 | C/T | 3.96 × 10−3 | 29.4 | 3 | 3/4 2 | Likely Pathogenic/Pathogenic |
| PMS2 3 | 172 | 12 | 0.021% | No dbSNP | 7 | 6043400 | T/C | . | 24.9 | 3 | 6 | - |
| BRCA1 | 344 | 17 | 0.071% | Not found | - | - | - | - | - | - | - | - |
| HOXB13 3 | 62 | 0 | Not found | - | - | - | - | - | - | - | - | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, G.; Catalano, C.; Bandapalli, O.R.; Paramasivam, N.; Chattopadhyay, S.; Schlesner, M.; Sijmons, R.; Hemminki, A.; Dymerska, D.; Lubinski, J.; et al. Cancer Predisposition Genes in Cancer-Free Families. Cancers 2020, 12, 2770. https://doi.org/10.3390/cancers12102770
Zheng G, Catalano C, Bandapalli OR, Paramasivam N, Chattopadhyay S, Schlesner M, Sijmons R, Hemminki A, Dymerska D, Lubinski J, et al. Cancer Predisposition Genes in Cancer-Free Families. Cancers. 2020; 12(10):2770. https://doi.org/10.3390/cancers12102770
Chicago/Turabian StyleZheng, Guoqiao, Calogerina Catalano, Obul Reddy Bandapalli, Nagarajan Paramasivam, Subhayan Chattopadhyay, Matthias Schlesner, Rolf Sijmons, Akseli Hemminki, Dagmara Dymerska, Jan Lubinski, and et al. 2020. "Cancer Predisposition Genes in Cancer-Free Families" Cancers 12, no. 10: 2770. https://doi.org/10.3390/cancers12102770
APA StyleZheng, G., Catalano, C., Bandapalli, O. R., Paramasivam, N., Chattopadhyay, S., Schlesner, M., Sijmons, R., Hemminki, A., Dymerska, D., Lubinski, J., Hemminki, K., & Försti, A. (2020). Cancer Predisposition Genes in Cancer-Free Families. Cancers, 12(10), 2770. https://doi.org/10.3390/cancers12102770