Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells

 ,
,  , , and
, , and
Abstract

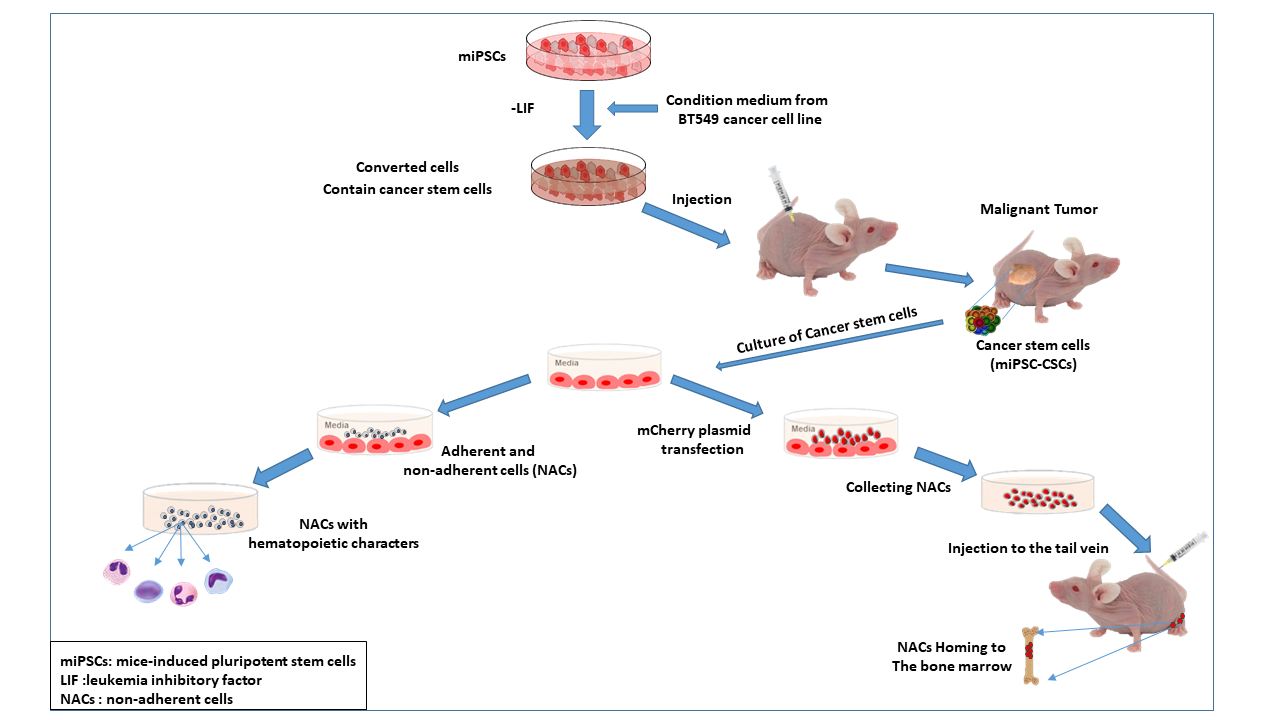
1. Introduction
2. Results
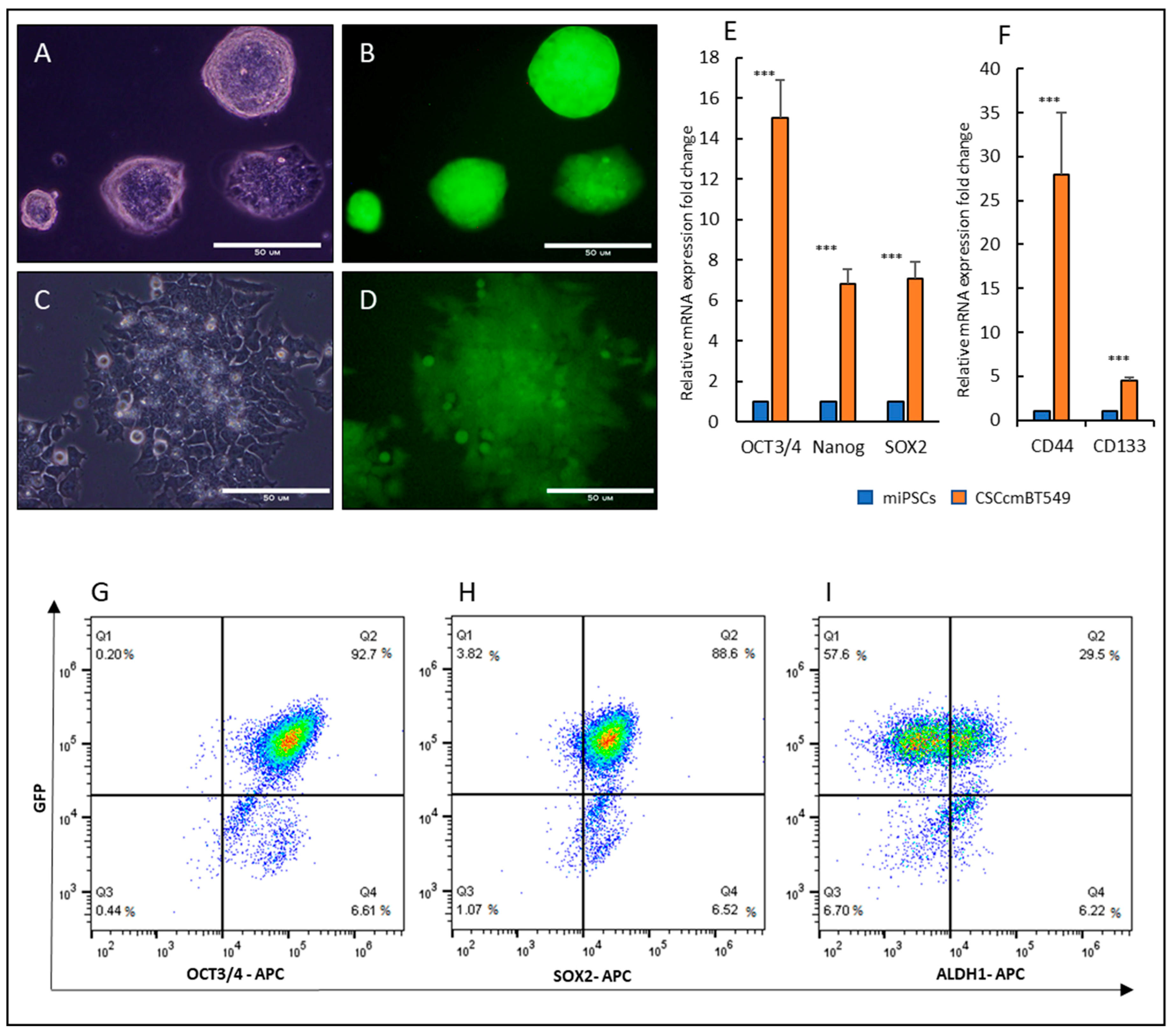
2.1. Cancer Stem Cells Converted from miPSCs
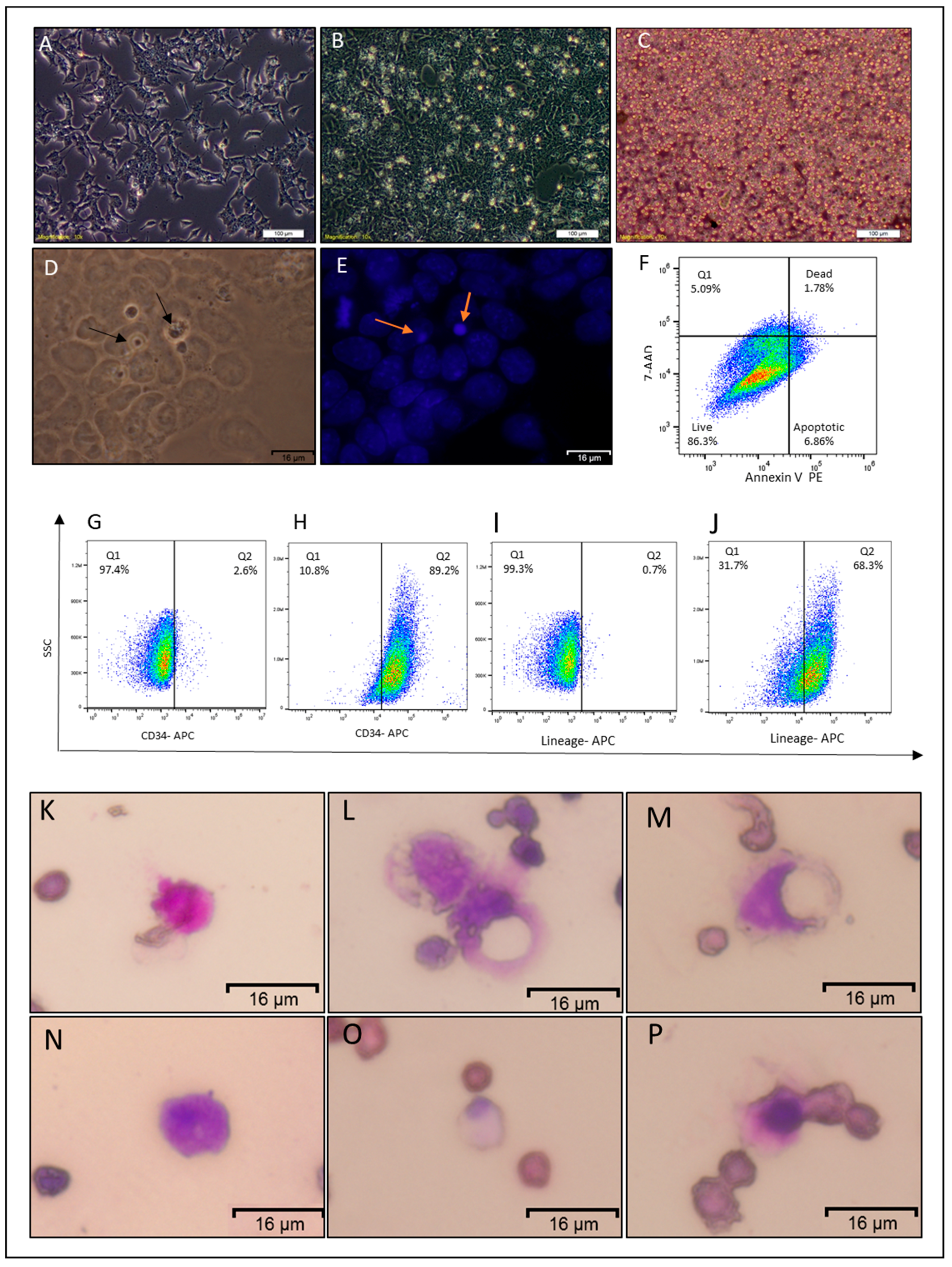
2.2. Non-Adherent Round Cells Emerging from CSCs
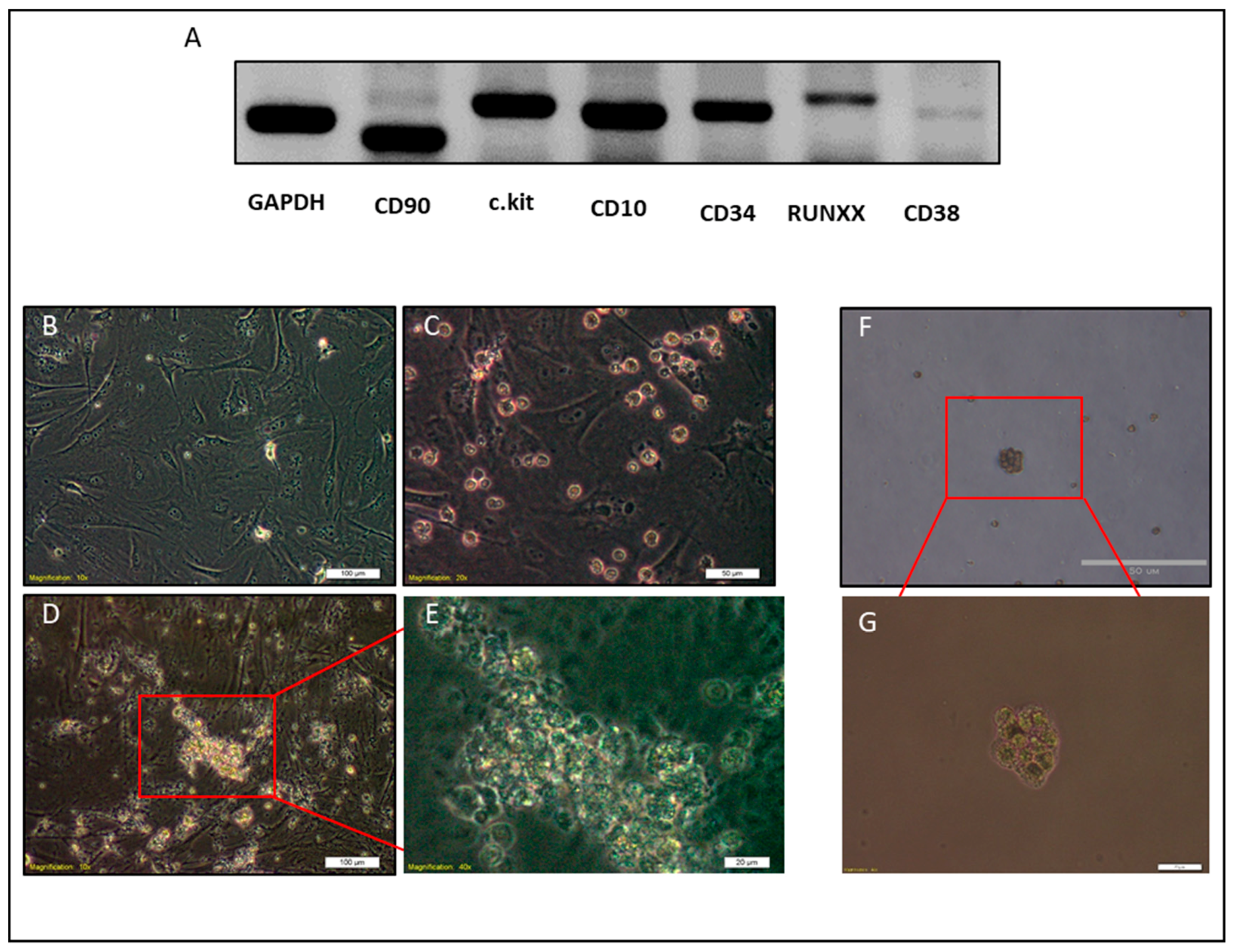
2.3. NACs Have Hematopoietic Cells Characteristics
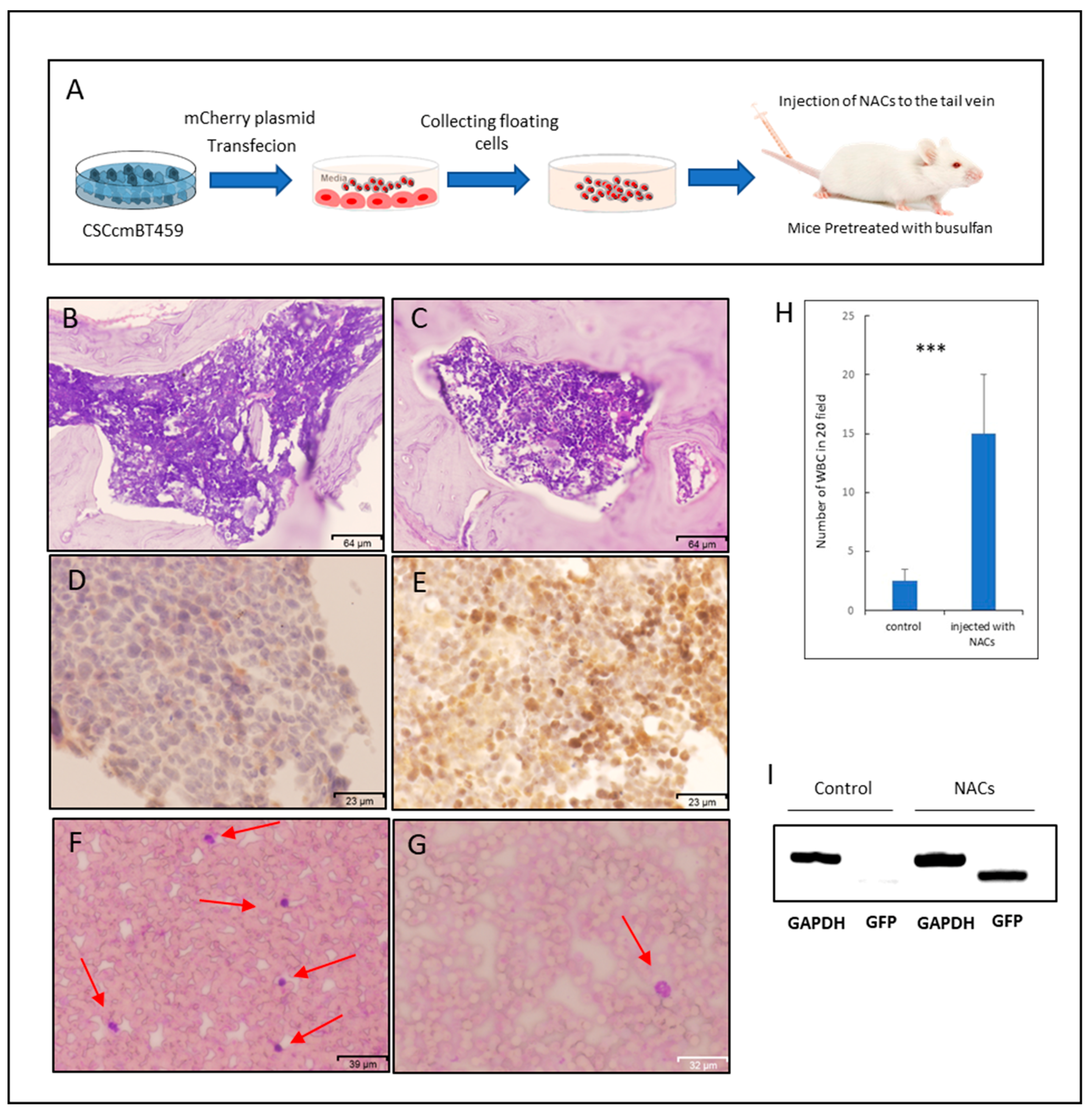
2.4. NACs Showed the Ability of Homing to Bone Marrow and Might Lead to Restoring Leukocytes in Busulfan-Conditioned Mice
2.5. Differentiation of CSCs into Macrophages
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Collection and Culture of NACs
4.3. RNA Extraction, cDNA Synthesis, and PCR
4.4. Flow Cytometry
4.5. Wright-Giemsa Staining
4.6. Immunofluorescence Staining
4.7. Clonogenic Assay
4.8. Introduction of mCherry Gene
4.9. Animal Experiments
4.10. DNA Extraction
4.11. Histological Analysis and Immunohistochemistry
4.12. Differentiation of CSCs into Macrophages
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.V.; Mielgo, A. Macrophages and Fibroblasts, Key Players in Cancer Chemoresistance. Front. Cell Dev. Biol. 2018, 6, 131. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 2017, 108, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, E.; Ko, E.; Ham, M.; Lee, H.M.; Kim, E.S.; Koh, M.; Lim, H.K.; Jung, J.; Park, S.Y.; et al. Tumor-associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1-alpha and MMP-9. Cancer Lett. 2018, 437, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco Targets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Tirino, V.; Desiderio, V.; Paino, F.; De Rosa, A.; Papaccio, F.; La Noce, M.; Laino, L.; De Francesco, F.; Papaccio, G. Cancer stem cells in solid tumors: An overview and new approaches for their isolation and characterization. FASEB J. 2013, 27, 13–24. [Google Scholar] [CrossRef]
- Mei, X.; Chen, Y.S.; Chen, F.R.; Xi, S.Y.; Chen, Z.P. Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro Oncol. 2017, 19, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 6838. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, W.; Fan, C.; Chen, X.; Lu, R.; Liu, Y.; Li, Y.; Shang, Y.; Yin, D.; Zhang, S.; Huang, Q.; et al. Endothelium originated from colorectal cancer stem cells constitute cancer blood vessels. Cancer Sci. 2017, 108, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, J.Y.; Ahn, J.H.; Ahn, S.H.; Gong, G. Tumor-associated lymphocytes predict response to neoadjuvant chemotherapy in breast cancer patients. J. Breast Cancer 2013, 16, 32–39. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Liu, J. Generation of erythroid cells from fibroblasts and cancer cells in vitro and in vivo. Cancer Lett. 2013, 333, 205–212. [Google Scholar] [CrossRef]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A Model of Cancer Stem Cells Derived from Mouse Induced Pluripotent Stem Cells. PLOS ONE 2012, 7, e33544. [Google Scholar] [CrossRef]
- Afify, S.M.; Hassan, G.; Osman, A.; Calle, A.S.; Nawara, H.M.; Zahra, M.H.; El-Ghlban, S.; Mansour, H.; Alam, M.J.; Abu Quora, H.A.; et al. Metastasis of Cancer Stem Cells Developed in the Microenvironment of Hepatocellular Carcinoma. Bioengineering 2019, 6, 73. [Google Scholar] [CrossRef]
- Calle, A.S.; Nair, N.; Oo, A.K.; Prieto-Vila, M.; Koga, M.; Khayrani, A.C.; Hussein, M.; Hurley, L.; Vaidyanath, A.; Seno, A.; et al. A new PDAC mouse model originated from iPSCs-converted pancreatic cancer stem cells (CSCcm). Am. J. Cancer Res. 2016, 6, 2799–2815. [Google Scholar]
- Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, T.; Liu, A.Y.; Ouyang, G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 2015, 6, 39550–39563. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Borovski, T.; De Sousa E Melo, F.; Vermeulen, L.; Medema, J.P. Cancer Stem Cell Niche: The Place to Be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef]
- Desai, A.; Yan, Y.; Gerson, S.L. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl. Med. 2019, 8, 75–81. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef]
- Lai, D.; Wang, F.; Chen, Y.; Wang, C.; Liu, S.; Lu, B.; Ge, X.; Guo, L. Human ovarian cancer stem-like cells can be efficiently killed by gammadelta T lymphocytes. Cancer Immunol. Immunother. 2012, 61, 979–989. [Google Scholar] [CrossRef]
- Todaro, M.; Orlando, V.; Cicero, G.; Caccamo, N.; Meraviglia, S.; Stassi, G.; Dieli, F. Chemotherapy sensitizes colon cancer initiating cells to Vgamma9Vdelta2 T cell-mediated cytotoxicity. PLoS ONE 2013, 8, e65145. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin. Transl. Med. 2018, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [PubMed]
- Seno, A.; Kasai, T.; Ikeda, M.; Vaidyanath, A.; Masuda, J.; Mizutani, A.; Murakami, H.; Ishikawa, T.; Seno, M. Characterization of Gene Expression Patterns among Artificially Developed Cancer Stem Cells Using Spherical Self-Organizing Map. Cancer Inform. 2016, 15, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Matic, I.Z.; Kolundzija, B.; Damjanovic, A.; Spasic, J.; Radosavljevic, D.; Dordic Crnogorac, M.; Grozdanic, N.; Juranic, Z.D. Peripheral White Blood Cell Subsets in Metastatic Colorectal Cancer Patients Treated with Cetuximab: The Potential Clinical Relevance. Front. Immunol. 2017, 8, 1886. [Google Scholar] [CrossRef]
- Rocca, Y.S.; Roberti, M.P.; Juliá, E.P.; Pampena, M.B.; Bruno, L.; Rivero, S.; Huertas, E.; Sánchez Loria, F.; Pairola, A.; Caignard, A.; et al. Phenotypic and Functional Dysregulated Blood NK Cells in Colorectal Cancer Patients Can Be Activated by Cetuximab Plus IL-2 or IL-15. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S.; Qu, J.; Zhang, M.; Li, Y.; Zhang, X.; Zhang, S. Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J. Cancer 2017, 8, 469–478. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Forward Primer | Reverse Primer |
|---|---|---|---|
| GAPDH | NM_008084 | AACGGCACAGTCAAGGCCGA | ACCCTTTTGGCTCCACCCTT |
| Nanog | NM_028016.3 | AGGGTCTGCTACTGAGATGCTCTG | CAACCACTGGTTTTTCTGCCACCG |
| OCT3/4 | NM_013633.3 | TCTTTCCACCAGGCCCCCGGCTC | TGCGGGCGGACATGGGGAGATCC |
| SOX2 | NM_011443.4 | TAGAGCTAGACTCCGGGCGATGA | TTGCCTTAAACAAGACCACGAAA |
| CD133 | NM_001163578.1 | CCTTGTGGTTCTTACGTTTGTTG | CGTTGACGACATTCTCAAGCTG |
| CD44 | NM_009851.2 | AGAAAAATGGCCGCTACAGTATC | TGCATGTTTCAAAACCCTTGC |
| CD90 | NM_009382.3 | TGCAGCTAGGGGAGTCCAGAAT | TCCAGGCGAAGGTTTTGGTT |
| c.kit | NM_021099.3 | CGGACAGCACCAAGCACATTTACTC | AACCATCACAGAAGCCAGAAGGACG |
| CD34 | NM_001111059.1 | TGCTGCATCTAAATAACTTGAC | AGGGATCCCAGAGGTAACTG |
| RUNX1 | NM_001111021.2 | CTGCCCATCGCTTTCAAGGTG | CTATGGTAGGTGGCAACTTGTGG |
| CD38 | NM_007646.5 | TGAGAGATCAGAACTGCCAGG | GTGTCCTCCAGGGTGAACAT |
| CD10 | NM_008604.4 | GCTAGAAGTCATTTTGAAAGATGTCCT | AGTGCCATATGTTTGATCCCAGT |
| CD11b | NM_001082960.1 | TACGTAATTGGGGTGGGAA | GTGCCCTCAATTGCAAAGAT |
| CD14 | NM_009841.3 | CTCTGTCCTTAAAGCGGCTTAC | GTTGCGGAGGTTCAAGATGTT |
| F4/80 | NM_010130.4 | CACCGGTATAGACAAGACTGACA | TCTCACCATCAGGAAGAGCA |
| CD68 | NM_009853.1 | ACTTCGGGCCATGTTTCTCT | GCTGGTAGGTTGATTGTCGT |
| GFP | GACAAGCAGAAGAACGGCATCAAGG | CTCAGGTAGTGGTTGTCGGGCAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, G.; Afify, S.M.; Nair, N.; Kumon, K.; Osman, A.; Du, J.; Mansour, H.; Abu Quora, H.A.; Nawara, H.M.; Satoh, A.; et al. Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells. Cancers 2020, 12, 82. https://doi.org/10.3390/cancers12010082
Hassan G, Afify SM, Nair N, Kumon K, Osman A, Du J, Mansour H, Abu Quora HA, Nawara HM, Satoh A, et al. Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells. Cancers. 2020; 12(1):82. https://doi.org/10.3390/cancers12010082
Chicago/Turabian StyleHassan, Ghmkin, Said M. Afify, Neha Nair, Kazuki Kumon, Amira Osman, Juan Du, Hager Mansour, Hagar A Abu Quora, Hend M Nawara, Ayano Satoh, and et al. 2020. "Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells" Cancers 12, no. 1: 82. https://doi.org/10.3390/cancers12010082
APA StyleHassan, G., Afify, S. M., Nair, N., Kumon, K., Osman, A., Du, J., Mansour, H., Abu Quora, H. A., Nawara, H. M., Satoh, A., Zahra, M. H., Okada, N., Seno, A., & Seno, M. (2020). Hematopoietic Cells Derived from Cancer Stem Cells Generated from Mouse Induced Pluripotent Stem Cells. Cancers, 12(1), 82. https://doi.org/10.3390/cancers12010082