Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
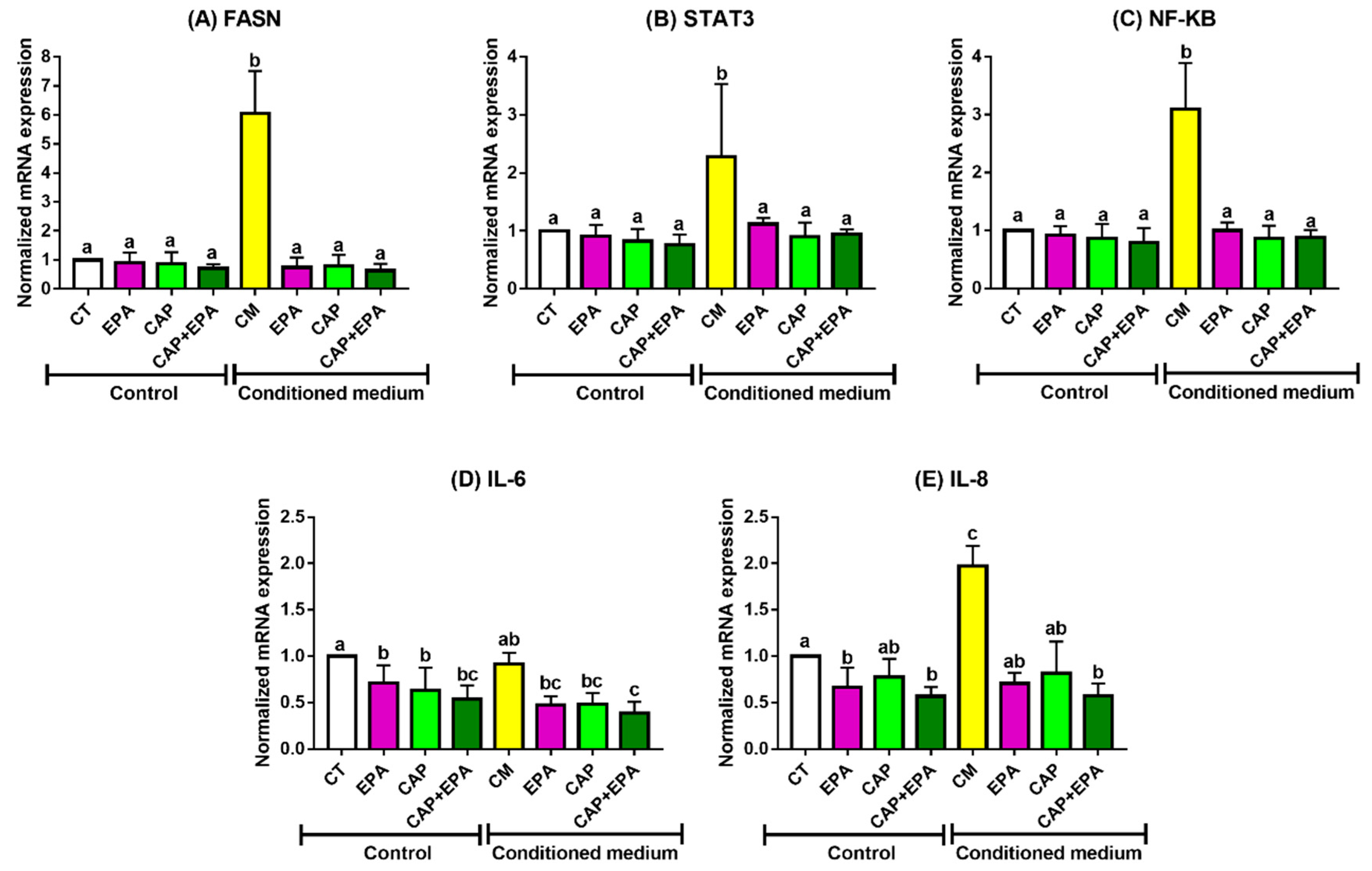
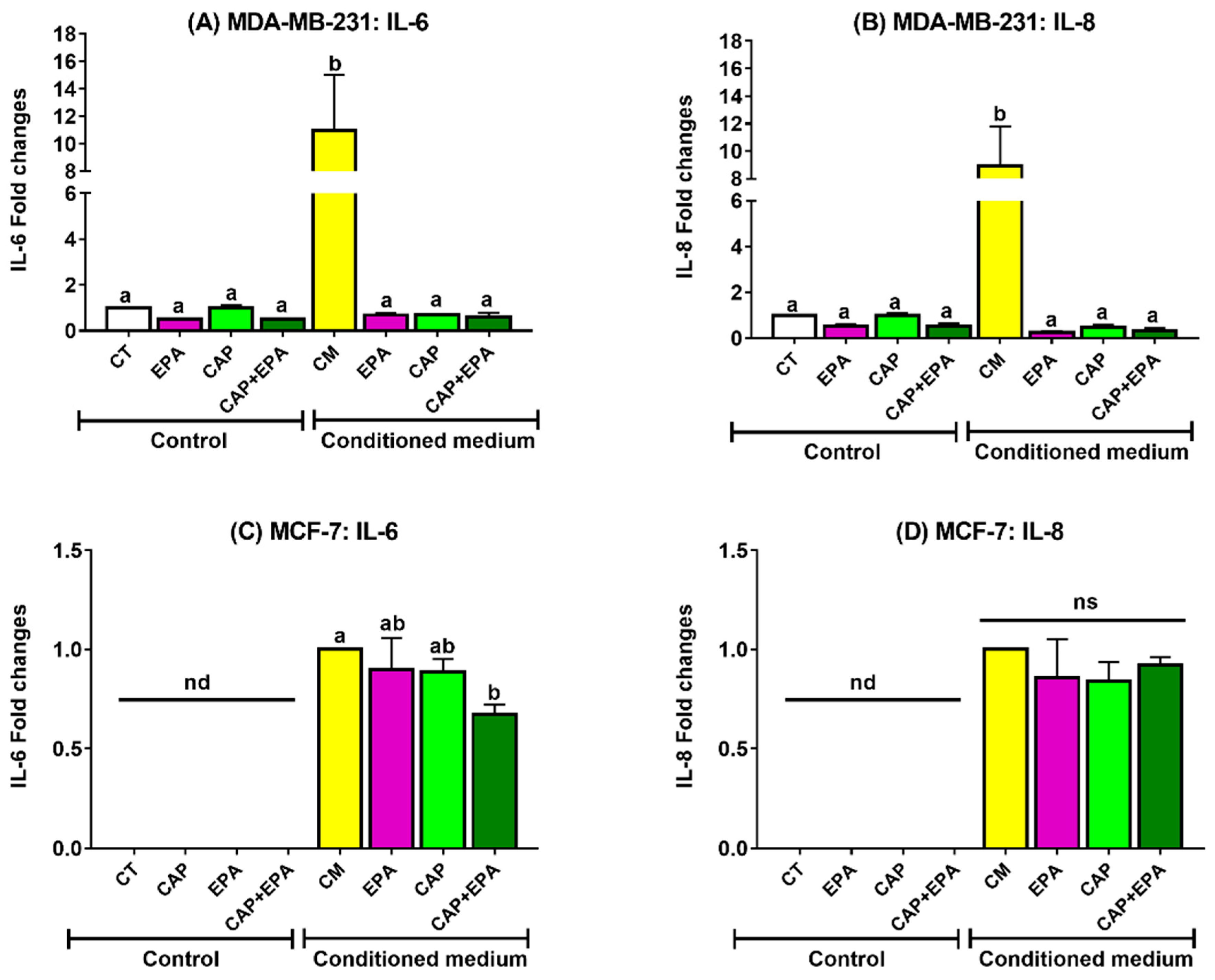
2.1. Effect of Captopril and EPA on Markers of Fatty Acid Synthesis and Inflammation in BC Cells and Role of Human Adipocyte-Conditioned Media (CM)
2.2. Combined Effect of Captopril and EPA on Breast Cancer Cell Migration in Response to Treatment with Adipocyte CM, Measured by a Wound Healing Assay
3. Discussion
4. Materials and Methods
4.1. Cell Culture Experiments
4.2. Treatment with ACE Inhibitor, Captopril, and Eicosapentaenoic Acid for Conditioned Medium Experiments
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. RNA Isolation and Real-Time Quatitative Polymerase Chain Reaction (RT-qPCR)
- IL-6 (5′-AGACAGCCACTCACCTCTTCAG-3′, 5′-TTTCTGCCAGTGCCTCTTTGC-3′),
- IL-8 (5′-AGGACAAGAGCCAGGAAGAA-3′, 5′-GGGTGGAAAGGTTTGGAGTATG-3′),
- NF-κB (5′-ATGGCTTCTATGAGGCTGAG-3′, 5′-GTTGTTGTTGGTCTGGATGC-3′),
- STAT3 (5′-AGAAGGACATCAGCGGTAAGA-3′, 5′-GGATAGAGATAGACCAGTGGAGAC-3′),
- FASN (5′-TCGTGGGCTACAGCATGGT-3′, 5′-GCCCTCTGAAGTCGAAGAAGAA-3X),
- 18S (5′-CTACCACATCCAAGGAAGCA-3′, 5’-TTTTTCGTCACTACCTCCCCG-3′), and
- TBP (5′-ATGGTGGTGTTGTGAGAAGATG-3′, 5′-CAGATAGCAGCACGGTATGAG-3′).
4.5. Wound Healing Assay
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Agt | Angiotensinogen |
| Ang I | Angiotensin I |
| Ang II | Angiotensin II |
| ACE | Angiotensin-converting enzyme |
| ACE-I | Angiotensin-converting enzyme inhibitor |
| ARB | Angiotensin receptor type I blocker |
| AT1R | Angiotensin receptor type I |
| AT2R | Angiotensin receptor type II |
| ATCC | American Type Culture Collection |
| BC | Breast cancer |
| CAA | Cancer-associated adipocytes |
| CAP | Captopril |
| CM | Conditioned medium |
| CVD | Cardiovascular disease |
| DHA | Docosahexaenoic acid |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| ELISA | Enzyme-linked immunosorbent assay |
| EPA | Eicosapentaenoic acid |
| FBS | Fetal bovine serum |
| FASN | Fatty acid synthase |
| ER | Estrogen receptor |
| ER+ | Estrogen receptor positive |
| ER- | Estrogen receptor negative |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HER2+ | Human epidermal growth factor receptor-2 positive |
| HMSC | Human mesenchymal stem cells |
| HUVEC | Human umbilical vein endothelial cells |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MMP | Matrix metalloproteinase |
| n-3 PUFA | Omega-3 polyunsaturated fatty acid |
| NF-κB | Nuclear factor kappa B |
| PBS | Phosphate-buffered saline |
| PR | Progesterone receptor |
| RAS | Renin–angiotensin system |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumor-associated macrophage |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| VEGF | Vascular endothelial growth factor |
References
- Stewart, B.; Wild, C.P. World Cancer Report 2014; World Health Organization (WHO): Geneva, Switzerland, 2017. [Google Scholar]
- Carmichael, A.R. Obesity as a risk factor for development and poor prognosis of breast cancer. BJOG Int. J. Obstet. Gynaecol. 2006, 113, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Schatzkin, A.; Lacey, J.V.; Albanes, D.; Ballard-Barbash, R.; Adams, K.F.; Kipnis, V.; Mouw, T.; Hollenbeck, A.R.; Leitzmann, M.F. Adiposity, Adult Weight Change, and Postmenopausal Breast Cancer Risk. Arch. Intern. Med. 2007, 167, 2091. [Google Scholar] [CrossRef] [PubMed]
- Simone, V.; D’avenia, M.; Argentiero, A.; Felici, C.; Rizzo, F.M.; De Pergola, G.; Silvestris, F. Obesity and breast cancer: Molecular interconnections and potential clinical applications. Oncologist 2016, 21, 404–417. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 421–449. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Wang, Y.; Kuhajda, F.P.; Li, J.N.; Pizer, E.S.; Han, W.F.; Sokoll, L.J.; Chan, D.W. Fatty acid synthase (FAS) expression in human breast cancer cell culture supernatants and in breast cancer patients. Cancer Lett. 2001, 167, 99–104. [Google Scholar] [CrossRef]
- Berndt, J.; Kovacs, P.; Ruschke, K.; Kloting, N.; Fasshauer, M.; Schon, M.R.; Korner, A.; Stumvoll, M.; Bluher, M. Fatty acid synthase gene expression in human adipose tissue: Association with obesity and type 2 diabetes. Diabetologia 2007, 50, 1472–1480. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Associations between obesity and cancer: The role of fatty acid synthase. J. Natl. Cancer Inst. 2012, 104, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, P.; Combs, T.P.; Shah, S.J.; Gouon-Evans, V.; Pollard, J.W.; Albanese, C.; Flanagan, L.; Tenniswood, M.P.; Guha, C.; Lisanti, M.P.; et al. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 2003, 22, 6408–6423. [Google Scholar] [CrossRef] [PubMed]
- Kalupahana, N.S.; Moustaid-Moussa, N. The adipose tissue renin-angiotensin system and metabolic disorders: A review of molecular mechanisms. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 379–390. [Google Scholar] [CrossRef]
- Jing, F.; Mogi, M.; Horiuchi, M. Role of renin–angiotensin–aldosterone system in adipose tissue dysfunction. Mol. Cell. Endocrinol. 2013, 378, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, L.; Menikdiwela, K.; LeMieux, M.; Dufour, J.M.; Kaur, G.; Kalupahana, N.; Moustaid-Moussa, N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim. et Biophys. Acta Mol. Basis Dis. 2017, 1863, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Namazi, S.; Rostami-Yalmeh, J.; Sahebi, E.; Jaberipour, M.; Razmkhah, M.; Hosseini, A. The role of captopril and losartan in prevention and regression of tamoxifen-induced resistance of breast cancer cell line MCF-7: An in vitro study. Biomed. Pharmacother. 2014, 68, 565–571. [Google Scholar] [CrossRef]
- Muscella, A.; Greco, S.; Elia, M.G.; Storelli, C.; Marsigliante, S. Angiotensin II stimulation of Na+/K+ATPase activity and cell growth by calcium-independent pathway in MCF-7 breast cancer cells. J. Endocrinol. 2002, 173, 315–323. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616. [Google Scholar] [CrossRef]
- Rodrigues-Ferreira, S.; Nahmias, C. G-protein coupled receptors of the renin-angiotensin system: New targets against breast cancer? Front. Pharmacol. 2015, 6, 24. [Google Scholar] [CrossRef]
- Ni, H.; Rui, Q.; Zhu, X.; Yu, Z.; Gao, R.; Liu, H. Antihypertensive drug use and breast cancer risk: A meta-analysis of observational studies. Oncotarget 2017, 8, 62545. [Google Scholar] [CrossRef]
- Ulu, A.; Harris, T.R.; Morisseau, C.; Miyabe, C.; Inoue, H.; Schuster, G.; Dong, H.; Iosif, A.M.; Liu, J.Y.; Weiss, R.H.; et al. Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 2013, 62, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Fabian, C.J.; Kimler, B.F.; Hursting, S.D. Omega-3 fatty acids for breast cancer prevention and survivorship. Breast Cancer Res. 2015, 17, 62. [Google Scholar] [CrossRef]
- Kalupahana, N.S.; Claycombe, K.; Newman, S.J.; Stewart, T.; Siriwardhana, N.; Matthan, N.; Lichtenstein, A.H.; Moustaid-Moussa, N. Eicosapentaenoic Acid Prevents and Reverses Insulin Resistance in High-Fat Diet-Induced Obese Mice via Modulation of Adipose Tissue Inflammation. J. Nutr. 2010, 140, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Kalupahana, N.S.; Claycombe, K.J.; Moustaid-Moussa, N. (n-3) Fatty Acids Alleviate Adipose Tissue Inflammation and Insulin Resistance: Mechanistic Insights. Adv. Nutr. 2011, 2, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Al-Jawadi, A.; Moussa, H.; Ramalingam, L.; Dharmawardhane, S.; Gollahon, L.; Gunaratne, P.; Layeequr Rahman, R.; Moustaid-Moussa, N. Protective properties of n-3 fatty acids and implications in obesity-associated breast cancer. J. Nutr. Biochem. 2018, 53, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al-Jawadi, A.; Rasha, F.; Ramalingam, L.; Alhaj, S.; Moussa, H.; Gollahon, L.; Dharmawardhane, S.; Moustaid-Moussa, N. Protective effects of eicosapentaenoic acid in adipocyte-breast cancer cell cross talk. J. Nutr. Biochem. 2020, 75, 108244. [Google Scholar] [CrossRef]
- Freund, A.; Jolivel, V.; Durand, S.; Kersual, N.; Chalbos, D.; Chavey, C.; Vignon, F.; Lazennec, G. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene 2004, 23, 6105–6114. [Google Scholar] [CrossRef]
- Siriwardhana, N.; Kalupahana, N.S.; Fletcher, S.; Xin, W.; Claycombe, K.J.; Quignard-Boulange, A.; Zhao, L.; Saxton, A.M.; Moustaid-Moussa, N. n-3 and n-6 polyunsaturated fatty acids differentially regulate adipose angiotensinogen and other inflammatory adipokines in part via NF-κB-dependent mechanisms. J. Nutr. Biochem. 2012, 23, 1661–1667. [Google Scholar] [CrossRef]
- Brinton, E.A.; Mason, R.P. Prescription omega-3 fatty acid products containing highly purified eicosapentaenoic acid (EPA). Lipids Health Dis. 2017, 16, 23. [Google Scholar] [CrossRef]
- Superko, H.R.; Superko, A.R.; Lundberg, G.P.; Margolis, B.; Garrett, B.C.; Nasir, K.; Agatston, A.S. Omega-3 Fatty Acid Blood Levels Clinical Significance Update. Curr. Cardiovasc. Risk Rep. 2014, 8, 407. [Google Scholar] [CrossRef]
- Itakura, H.; Yokoyama, M.; Matsuzaki, M.; Saito, Y.; Origasa, H.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Kita, T.; et al. Relationships between plasma fatty acid composition and coronary artery disease. J. Atheroscler. Thromb. 2011, 18, 99–107. [Google Scholar] [CrossRef]
- Braeckman, R.A.; Stirtan, W.G.; Soni, P.N. Pharmacokinetics of Eicosapentaenoic Acid in Plasma and Red Blood Cells After Multiple Oral Dosing With Icosapent Ethyl in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2014, 3, 101–108. [Google Scholar] [CrossRef]
- Song, J.; Li, C.; Lv, Y.; Zhang, Y.; Amakye, W.K.; Mao, L. DHA increases adiponectin expression more effectively than EPA at relative low concentrations by regulating PPARγ and its phosphorylation at Ser273 in 3T3-L1 adipocytes. Nutr. Metab. 2017, 14, 52. [Google Scholar] [CrossRef]
- Mansara, P.P.; Deshpande, R.A.; Vaidya, M.M.; Kaul-Ghanekar, R. Differential Ratios of Omega Fatty Acids (AA/EPA+DHA) Modulate Growth, Lipid Peroxidation and Expression of Tumor Regulatory MARBPs in Breast Cancer Cell Lines MCF7 and MDA-MB-231. PLoS ONE 2015, 10, e0136542. [Google Scholar] [CrossRef]
- Cunha, J.P. Consumer_Captopril_Capoten. Available online: https://www.rxlist.com/consumer_captopril_capoten/drugs-condition.htm (accessed on 3 January 2020).
- Small, W., Jr.; James, J.L.; Moore, T.D.; Fintel, D.J.; Lutz, S.T.; Movsas, B.; Suntharalingam, M.; Garces, Y.I.; Ivker, R.; Moulder, J.; et al. Utility of the ACE Inhibitor Captopril in Mitigating Radiation-associated Pulmonary Toxicity in Lung Cancer: Results From NRG Oncology RTOG 0123. Am. J. Clin. Oncol. 2018, 41, 396–401. [Google Scholar] [CrossRef]
- Guglin, M.; Munster, P.; Fink, A.; Krischer, J. Lisinopril or Coreg CR in reducing cardiotoxicity in women with breast cancer receiving trastuzumab: A rationale and design of a randomized clinical trial. Am. Heart J. 2017, 188, 87–92. [Google Scholar] [CrossRef]
- Onoyama, K.; Hirakata, H.; Iseki, K.; Fujimi, S.; Omae, T.; Kobayashi, M.; Kawahara, Y. Blood concentration and urinary excretion of captopril (SQ 14,225) in patients with chronic renal failure. Hypertension 1981, 3, 456–459. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Sagaradze, G.; Grigorieva, O.; Nimiritsky, P.; Basalova, N.; Kalinina, N.; Akopyan, Z.; Efimenko, A. Conditioned Medium from Human Mesenchymal Stromal Cells: Towards the Clinical Translation. Int. J. Mol. Sci. 2019, 20, 1656. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.C.; Church, F.C. Mature breast adipocytes promote breast cancer cell motility. Exp. Mol. Pathol. 2012, 92, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Colomer, R.; Brunet, J.; Lupu, R.; Menendez, J.A. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008, 41, 59–85. [Google Scholar] [CrossRef] [PubMed]
- Alwarawrah, Y.; Hughes, P.; Loiselle, D.; Carlson, D.A.; Darr, D.B.; Jordan, J.L.; Xiong, J.; Hunter, L.M.; Dubois, L.G.; Thompson, J.W.; et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem. Biol. 2016, 23, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, L.; Costanzo, C.; Merola, M.; Bianchini, E.; Furia, A.; Carsana, A.; Palmieri, M. Nuclear factor kappa B (NF-kappa B), nuclear factor interleukin-6 (NFIL-6 or C/EBP beta) and nuclear factor interleukin-6 beta (NFIL6-beta or C/EBP delta) are not sufficient to activate the endogenous interleukin-6 gene in the human breast carcinoma cell line MCF-7. Comparative analysis with MDA-MB-231 cells, an interleukin-6-expressing human breast carcinoma cell line. Eur. J. Biochem. 1996, 239, 624–631. [Google Scholar] [PubMed]
- Chavey, C.; Muhlbauer, M.; Bossard, C.; Freund, A.; Durand, S.; Jorgensen, C.; Jobin, C.; Lazennec, G. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-kappaB pathway in breast cancer. Mol. Pharmacol. 2008, 74, 1359–1366. [Google Scholar] [CrossRef]
- Bravata, V.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Russo, G.; Di Maggio, F.M.; Augello, G.; Lio, D.; Gilardi, M.C. Cytokine profile of breast cell lines after different radiation doses. Int. J. Radiat. Biol. 2017, 93, 1217–1226. [Google Scholar] [CrossRef]
- Trebble, T.; Arden, N.K.; Stroud, M.A.; Wootton, S.A.; Burdge, G.C.; Miles, E.A.; Ballinger, A.B.; Thompson, R.L.; Calder, P.C. Inhibition of tumour necrosis factor-a and interleukin-6 production by mononuclear cells following dietary fish-oil supplementation in healthy men and response to antioxidant co-supplementation. Br. J. Nutr. 2003, 90, 405–412. [Google Scholar] [CrossRef]
- Duvall, M.G.; Levy, B. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2016, 785, 144–155. [Google Scholar] [CrossRef]
- Illan-Cabeza, N.A.; Jimenez-Pulido, S.B.; Hueso-Urena, F.; Ramirez-Exposito, M.J.; Sanchez-Sanchez, P.; Martinez-Martos, J.M.; Moreno-Carretero, M.N. Effects on estrogen-dependent and triple negative breast cancer cells growth of Ni(II), Zn(II) and Cd(II) complexes with the Schiff base derived from pyridine-2-carboxaldehyde and 5,6-diamino-1,3-dimethyluracil explored through the renin-angiotensin system (RAS)-regulating aminopeptidases. J. Inorg. Biochem. 2018, 185, 52–62. [Google Scholar] [CrossRef]
- Brown, I.; Lee, J.; Sneddon, A.A.; Cascio, M.G.; Pertwee, R.G.; Wahle, K.W.; Rotondo, D.; Heys, S.D. Anticancer effects of n-3 EPA and DHA and their endocannabinoid derivatives on breast cancer cell growth and invasion. Prostaglandins Leukot. Essent. Fat. Acids 2019. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.H.; Leung, W.H.; Pang, Y.J.; Kuo, L.W.; Hsu, H.H. EPA significantly improves anti-EGFR targeted therapy by regulating miR-378 expression in colorectal cancer. Oncol. Lett. 2018, 16, 6188–6194. [Google Scholar] [CrossRef] [PubMed]
- Niazi, Z.R.; Silva, G.C.; Ribeiro, T.P.; Leon-Gonzalez, A.J.; Kassem, M.; Mirajkar, A.; Alvi, A.; Abbas, M.; Zgheel, F.; Schini-Kerth, V.B.; et al. EPA:DHA 6:1 prevents angiotensin II-induced hypertension and endothelial dysfunction in rats: Role of NADPH oxidase- and COX-derived oxidative stress. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2017, 40, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Stephen Lee, K.S.; Miyabe, C.; Yang, J.; Hammock, B.G.; Dong, H.; Hammock, B.D. An omega-3 epoxide of docosahexaenoic acid lowers blood pressure in angiotensin-II-dependent hypertension. J. Cardiovasc. Pharmacol. 2014, 64, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Krusche, B.; Arend, J.; Efferth, T. Synergistic inhibition of angiogenesis by artesunate and captopril in vitro and in vivo. Evid. Based Complement. Alternat. Med. 2013, 2013, 454783. [Google Scholar] [CrossRef]
- Miguel-Carrasco, J.L.; Zambrano, S.; Blanca, A.J.; Mate, A.; Vazquez, C.M. Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. J. Inflamm. 2010, 7, 21. [Google Scholar] [CrossRef]
- Lee, M.J.; Fried, S.K. Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods Enzymol. 2014, 538, 49–65. [Google Scholar] [CrossRef]
- Wortman, P.; Miyazaki, Y.; Kalupahana, N.S.; Kim, S.; Hansen-Petrik, M.; Saxton, A.M.; Claycombe, K.J.; Voy, B.H.; Whelan, J.; Moustaid-Moussa, N. n3 and n6 polyunsaturated fatty acids differentially modulate prostaglandin E secretion but not markers of lipogenesis in adipocytes. Nutr. Metab. 2009, 6, 5. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasha, F.; Kahathuduwa, C.; Ramalingam, L.; Hernandez, A.; Moussa, H.; Moustaid-Moussa, N. Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration. Cancers 2020, 12, 220. https://doi.org/10.3390/cancers12010220
Rasha F, Kahathuduwa C, Ramalingam L, Hernandez A, Moussa H, Moustaid-Moussa N. Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration. Cancers. 2020; 12(1):220. https://doi.org/10.3390/cancers12010220
Chicago/Turabian StyleRasha, Fahmida, Chanaka Kahathuduwa, Latha Ramalingam, Arelys Hernandez, Hanna Moussa, and Naima Moustaid-Moussa. 2020. "Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration" Cancers 12, no. 1: 220. https://doi.org/10.3390/cancers12010220
APA StyleRasha, F., Kahathuduwa, C., Ramalingam, L., Hernandez, A., Moussa, H., & Moustaid-Moussa, N. (2020). Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration. Cancers, 12(1), 220. https://doi.org/10.3390/cancers12010220