Precision Medicine in Soft Tissue Sarcoma Treatment


Abstract
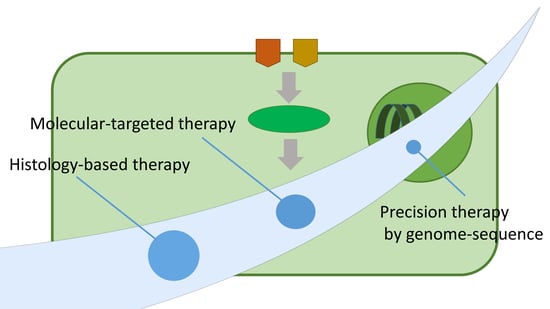
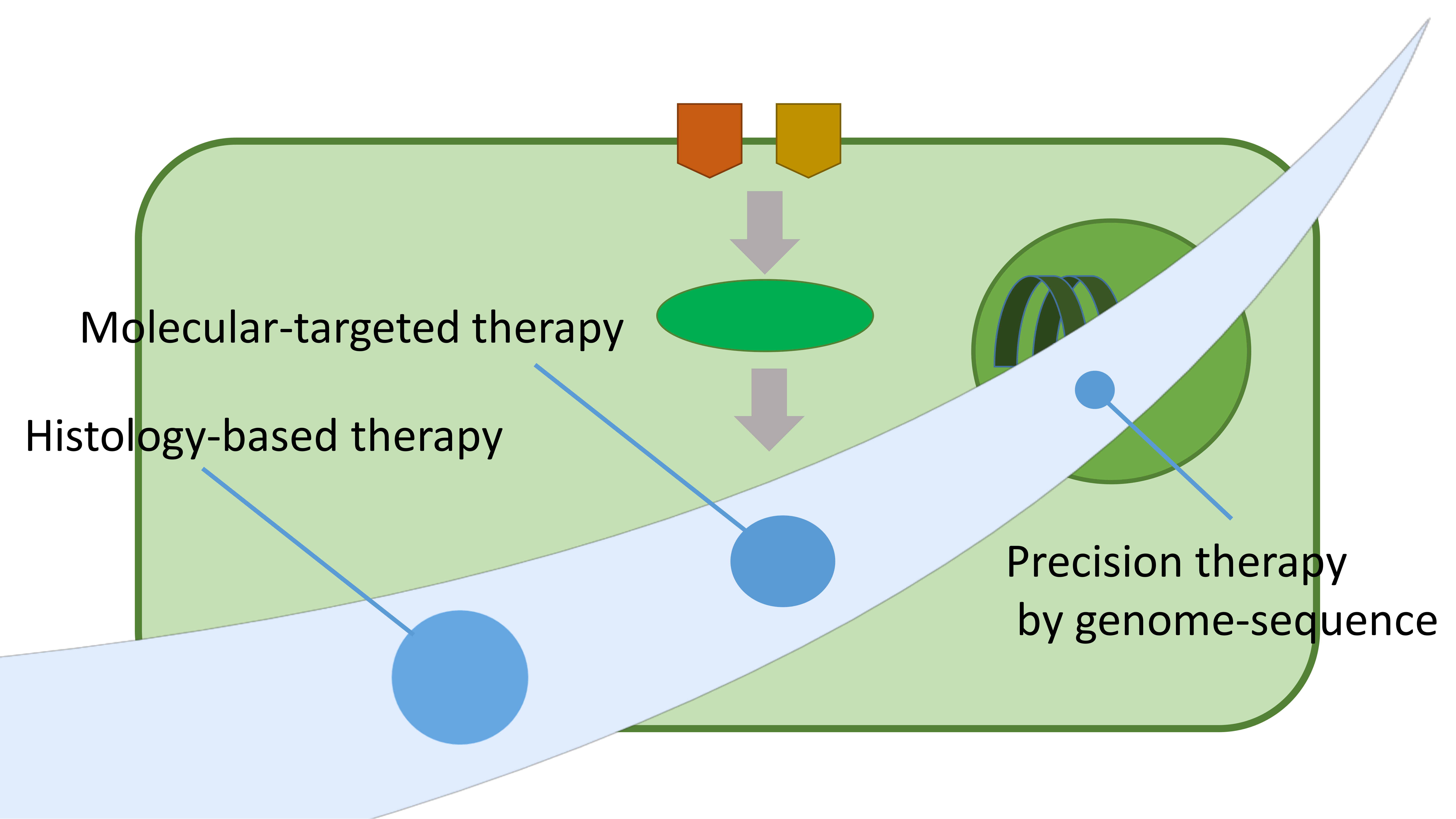
1. Introduction
2. The Limitations of Incorporating STS Patients Regardless of Their Histological Subtypes
3. Histology-Based Chemotherapy Investigations Based on Clinical Data
4. Investigating Molecular Targeted Therapies for STS Patients
5. Whole-Genome Sequencing for Precision Medicine for STS Patients
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALK | anaplastic lymphoma kinase |
| ASPS | alveolar soft part sarcoma |
| CDK4 | cyclin-dependent kinase 4 |
| EMA | European Medicines Agency |
| EORTC | European Organisation for Research and Treatment of Cancer |
| FDA | Food and Drug Administration |
| GIST | gastrointestinal stromal tumor |
| IGF1-R | insulin-like growth factor 1 receptor |
| IMT | inflammatory myofibroblastic tumor |
| MDM2 | murine double minute 2 |
| MSI | microsatellite instability |
| MPNST | malignant peripheral nerve sheath tumor |
| mTOR | mammalian target of rapamycin |
| NGS | next generation sequencing |
| NTRK | neurotrophic receptor kinase |
| OS | overall survival |
| PDGFR | platelet-derived growth factor receptor |
| PD-L1 | programmed death ligand-1 |
| PEComa | perivascular epithelioid cell tumor |
| PFS | progression-free survival |
| PI3K | phosphatidylinositol 3-kinase |
| STS | soft tissue sarcoma |
| TKI | tyrosine kinase inhibitor |
| TMB | tumor mutation burden |
| UPS | undifferentiated pleomorphic sarcoma |
| VEGF | vascular endothelial growth factor |
| XPO1 | exportin-1 |
References
- DeVita, V.T., Jr.; Lawrence, T.S.; Rosenberg, S.A. DeVita, Hellman, and Rosenberg’s Cancer Principles & Practice of Oncology, 11th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2018. [Google Scholar]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; International Agency Research on Cancer: Lyon, France, 2013; ISBN 978-92-832-2434-1. [Google Scholar]
- Nangalia, J.; Campbell, P.J. Genome sequencing during a patient’s journey through cancer. N. Engl. J. Med. 2019, 381, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Billingham, L.; Malottki, K.; Steven, N. Research methods to change clinical practice for patients with rare cancers. Lancet Oncol. 2016, 17, e70–e80. [Google Scholar] [CrossRef]
- Linch, M.; Miah, A.B.; Thway, K.; Judson, I.R.; Benson, C. Systemic treatment of soft-tissue sarcoma—Gold standard and novel therapies. Nat. Rev. Clin. Oncol. 2014, 11, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. European Organisation and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Ryan, C.W.; Merimsky, O.; Agulnik, M.; Blay, J.Y.; Schuetze, S.M.; Van Tine, B.A.; Jones, R.L.; Elias, A.D.; Choy, E.; Alcindor, T.; et al. PICASSO III: A Phase III, Placebo-controlled study of doxorubicin with or without palifosfamide in patients with metastatic soft tissue sarcoma. J. Clin. Oncol. 2016, 34, 3898–3905. [Google Scholar] [CrossRef]
- Tap, W.D.; Papai, Z.; Van Tine, B.A.; Attia, S.; Ganjoo, K.N.; Jones, R.L.; Schuetze, S.; Reed, D.; Chawla, S.P.; Riedel, R.F.; et al. Doxorubicin plus evofosfamide versus doxorubicin alone in locally advanced, unresectable or metastatic soft-tissue sarcoma (TH CR-406/SARC021): An international, multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2017, 18, 1089–1103. [Google Scholar] [CrossRef]
- Tap, W.D.; Wagner, A.J.; Papai, Z.; Ganjoo, K.N.; Yen, C.C.; Schöffski, P.; Razak, A.R.A.; Broto, J.M.; Spira, A.I.; Kawai, A.; et al. ANNOUNCE: A randomized, placebo (PBO)-controlled, double-blind, phase (ph) III trial of doxorubicin (dox) + olaratumab versus dox + PBO in patients (pts) with advanced soft tissue sarcomas (STS). J. Clin. Oncol. 2019, 36 (Suppl. 18). [Google Scholar] [CrossRef]
- Tap, W.D.; Jones, R.L.; Van Tine, B.A.; Chmielowski, B.; Elias, A.D.; Adkins, D.; Agulnik, M.; Cooney, M.M.; Livingston, M.B.; Pennock, G.; et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 2016, 388, 488–497. [Google Scholar] [CrossRef]
- Italiano, A. Olaratumab failure in sarcomas: What are the lessons learned? Eur. J. Cancer 2019, 117, 69–70. [Google Scholar] [CrossRef]
- Lorigan, P.; Verweij, J.; Papai, Z.; Rodenhuis, S.; Le Cesne, A.; Leahy, M.G.; Radford, J.A.; Van Glabbeke, M.M.; Kirkpatrick, A.; Hogendoorn, P.C.; et al. Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: A european organisation for research and treatment of cancer soft tissue and bone sarcoma group study. J. Clin. Oncol. 2007, 25, 3144–3150. [Google Scholar] [CrossRef]
- Seddon, B.; Strauss, S.J.; Whelan, J.; Leahy, M.; Woll, P.J.; Cowie, F.; Rothermundt, C.; Wood, Z.; Benson, C.; Ali, N.; et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): A randomised controlled phase 3 trial. Lancet Oncol. 2017, 18, 1397–1410. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing sarcoma: Current management and future approaches through collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Pappa, A.S.; Dirksen, U. Rhabdomyosarcoma, Ewing sarcoma, and other round cell sarcomas. J. Clin. Oncol. 2018, 36, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Beaino, E.; Araujo, D.M.; Lazar, A.J.; Lin, P.P. Synovial sarcoma: Advances in diagnosis and treatment identification of new biologic targets to improve multimodal therapy. Ann. Surg. Oncol. 2017, 24, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Eilber, F.C.; Brennan, M.F.; Eilber, F.R.; Eckardt, J.J.; Grobmyer, S.R.; Riedel, E.; Forscher, C.; Maki, R.G.; Singer, S. Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann. Surg. 2007, 246, 105–113. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ouali, M.; van Glabbeke, M.; Krarup-Hansen, A.; Rodenhuis, S.; Le Cesne, A.; Hogendoorn, P.C.; Verweij, J.; Blay, J.Y. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: An exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG). Eur. J. Cancer 2010, 46, 72–83. [Google Scholar] [CrossRef]
- Hensley, M.L.; Maki, R.; Venkatraman, E.; Geller, G.; Lovegren, M.; Aghajanian, C.; Sabbatini, P.; Tong, W.; Barakat, R.; Spriggs, D.R. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: Results of a phase II trial. J. Clin. Oncol. 2002, 20, 2824–2831. [Google Scholar] [CrossRef]
- Penel, N.; Bui, B.N.; Bay, J.O.; Cupissol, D.; Ray-Coquard, I.; Piperno-Neumann, S.; Kerbrat, P.; Fournier, C.; Taieb, S.; Jimenez, M.; et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: The ANGIOTAX Study. J. Clin. Oncol. 2008, 26, 5269–5274. [Google Scholar] [CrossRef]
- Penel, N.; Italiano, A.; Ray-Coquard, I.; Chaigneau, L.; Delcambre, C.; Robin, Y.M.; Bui, B.; Bertucci, F.; Isambert, N.; Cupissol, D.; et al. French Sarcoma Group (GSF/GETO). Metastatic angiosarcomas: Doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve the outcome. Ann. Oncol. 2012, 23, 517–523. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: Results of a Phase III randomized multicenter clinical trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; De Brauwer, A.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European Organisation for Research and Treatment of Cancer-soft Tissue and Bone Sarcoma Group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.L.; Chawla, S.; Patel, S.; von Mehren, M.; Hamm, J.; Kaiser, P.E.; Schuetze, S.; Li, J.; Aymes, A.; Demetri, G.D. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: Results of a worldwide expanded access program study. Ann. Oncol. 2013, 24, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Le Cesne, A.; Ray-Coquard, I.; Duffaud, F.; Chevreau, C.; Penel, N.; Bui Nguyen, B.; Piperno-Neumann, S.; Delcambre, C.; Rios, M.; Chaigneau, L.; et al. Trabectedin in patients with advanced soft tissue sarcoma: A retrospective national analysis of the French Sarcoma Group. Eur. J. Cancer 2015, 51, 742–750. [Google Scholar] [CrossRef]
- Barone, A.; Chi, D.C.; Theoret, M.R.; Chen, H.; He, K.; Kufrin, D.; Helms, W.S.; Subramaniam, S.; Zhao, H.; Patel, A.; et al. FDA approval summary: Trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin. Cancer Res. 2017, 23, 7448–7453. [Google Scholar] [CrossRef][Green Version]
- Le Cesne, A.; Cresta, S.; Maki, R.G.; Blay, J.Y.; Verweij, J.; Poveda, A.; Casali, P.G.; Balaña, C.; Schöffski, P.; Grosso, F.; et al. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer 2012, 48, 3036–3044. [Google Scholar] [CrossRef]
- Grosso, F.; Jones, R.L.; Demetri, G.D.; Judson, I.R.; Blay, J.Y.; Le Cesne, A.; Sanfilippo, R.; Casieri, P.; Collini, P.; Dileo, P.; et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar] [CrossRef]
- Blay, J.Y.; Leahy, M.G.; Nguyen, B.B.; Patel, S.R.; Hohenberger, P.; Santoro, A.; Staddon, A.P.; Penel, N.; Piperno-Neumann, S.; Hendifar, A.; et al. Randomised phase III trial of trabectedin versus doxorubicin-based chemotherapy as first-line therapy in translocation-related sarcomas. Eur. J. Cancer 2014, 50, 1137–1147. [Google Scholar] [CrossRef]
- Kawai, A.; Araki, N.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: A randomised, open-label, phase 2 study. Lancet Oncol. 2015, 16, 406–416. [Google Scholar] [CrossRef]
- Schöffski, P.; Ray-Coquard, I.L.; Cioffi, A.; Bui, N.B.; Bauer, S.; Hartmann, J.T.; Krarup-Hansen, A.; Grünwald, V.; Sciot, R.; Dumez, H.; et al. European Organisation for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group (STBSG). Activity of eribulin mesylate in patients with soft-tissue sarcoma: A phase 2 study in four independent histological subtypes. Lancet Oncol. 2011, 12, 1045–1052. [Google Scholar] [CrossRef]
- Demetri, G.D.; Schöffski, P.; Grignani, G.; Blay, J.Y.; Maki, R.G.; Van Tine, B.A.; Alcindor, T.; Jones, R.L.; D’Adamo, D.R.; Guo, M.; et al. Activity of eribulin in patients with advanced liposarcoma demonstrated in a subgroup analysis from a randomized phase III study of eribulin versus dacarbazine. J. Clin. Oncol. 2017, 35, 3433–3439. [Google Scholar] [CrossRef]
- Blay, J.Y.; Schöffski, P.; Bauer, S.; Krarup-Hansen, A.; Benson, C.; D’Adamo, D.R.; Jia, Y.; Maki, R.G. Eribulin versus dacarbazine in patients with leiomyosarcoma: Subgroup analysis from a phase 3, open-label, randomised study. Br. J. Cancer 2019, 120, 1026–1032. [Google Scholar] [CrossRef]
- Osgood, C.L.; Chuk, M.K.; Theoret, M.R.; Huang, L.; He, K.; Her, L.; Keegan, P.; Pazdur, R. FDA approval summary: Eribulin for patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen. Clin. Cancer Res. 2017, 23, 6384–6389. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.L.; Chawla, S.P.; Somaiah, N.; Staddon, A.P.; Skubitz, K.M.; Milhem, M.M.; Kaiser, P.E.; Portnoy, D.C.; Priebat, D.A.; Walker, M.S.; et al. Results of a prospective phase 2 study of pazopanib in patients with advanced intermediate-grade or high-grade liposarcoma. Cancer 2017, 123, 4640–4647. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Naito, Y.; Asano, N.; Maejima, A.; Endo, M.; Takahashi, S.; Megumi, Y.; Kawai, A. Interim results of a real-world observational study of eribulin in soft tissue sarcoma including rare subtypes. Jpn. J. Clin. Oncol. 2019, 49, 938–946. [Google Scholar] [CrossRef]
- Gronchi, A.; Ferrari, S.; Quagliuolo, V.; Broto, J.M.; Pousa, A.L.; Grignani, G.; Basso, U.; Blay, J.Y.; Tendero, O.; Beveridge, R.D.; et al. Histology-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft tissue sarcomas (ISG-STS 1001): An international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol. 2017, 18, 812–822. [Google Scholar] [CrossRef]
- Reichardt, P. The Story of Imatinib in GIST—A journey through the development of a targeted therapy. Oncol. Res. Treat. 2018, 41, 472–477. [Google Scholar] [CrossRef]
- Plaat, B.E.; Hollema, H.; Molenaar, W.M.; Torn Broers, G.H.; Pijpe, J.; Mastik, M.F.; Hoekstra, H.J.; van den Berg, E.; Scheper, R.J.; van der Graaf, W.T. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: Differences in clinical outcome and expression of multidrug resistance proteins. J. Clin. Oncol. 2000, 18, 3211–3220. [Google Scholar] [CrossRef]
- Joensuu, H.; Roberts, P.J.; Sarlomo-Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N. Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Brodowicz, T.; Mir, O.; Wallet, J.; Italiano, A.; Blay, J.Y.; Bertucci, F.; Eisterer, W.; Chevreau, C.; Piperno-Neumann, S.; Bompas, E.; et al. Efficacy and safety of regorafenib compared to placebo and to post-cross-over regorafenib in advanced non-adipocytic soft tissue sarcoma. Eur. J. Cancer 2018, 99, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Kollár, A.; Jones, R.L.; Stacchiotti, S.; Gelderblom, H.; Guida, M.; Grignani, G.; Steeghs, N.; Safwat, A.; Katz, D.; Duffaud, F.; et al. Pazopanib in advanced vascular sarcomas: An EORTC Soft Tissue and Bone Sarcoma Group (STBSG) retrospective analysis. Acta Oncol. 2017, 56, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Tamborini, E.; Marrari, A.; Brich, S.; Rota, S.A.; Orsenigo, M.; Crippa, F.; Morosi, C.; Gronchi, A.; Pierotti, M.A.; et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin. Cancer Res. 2009, 15, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Allen, D.; Monks, A.; Polley, E.C.; Hose, C.D.; Ivy, S.P.; Turkbey, I.B.; Lawrence, S.; Kinders, R.J.; Choyke, P.; et al. Cediranib for metastatic alveolar soft part sarcoma. J. Clin. Oncol. 2013, 31, 2296–2302. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.M.; Keam, B.; Kim, Y.J.; Paeng, J.C.; Moon, K.C.; Kim, D.W.; Heo, D.S. A phase II trial of pazopanib in patients with metastatic alveolar soft part sarcoma. Oncologist 2019, 24. [Google Scholar] [CrossRef]
- Agulnik, M.; Yarber, J.L.; Okuno, S.H.; von Mehren, M.; Jovanovic, B.D.; Brockstein, B.E.; Evens, A.M.; Benjamin, R.S. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann. Oncol. 2013, 24, 257–263. [Google Scholar] [CrossRef]
- Ray-Coquard, I.L.; Domont, J.; Tresch-Bruneel, E.; Bompas, E.; Cassier, P.A.; Mir, O.; Piperno-Neumann, S.; Italiano, A.; Chevreau, C.; Cupissol, D.; et al. Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: A randomized phase II trial. J. Clin. Oncol. 2015, 33, 2797–2802. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Morden, J.P.; Kilburn, L.; Leahy, M.; Benson, C.; Bhadri, V.; Campbell-Hewson, Q.; Cubedo, R.; Dangoor, A.; Fox, L.; et al. Cediranib in patients with alveolar soft-part sarcoma (CASPS): A double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef]
- Olmos, D.; Tan, D.S.; Jones, R.L.; Judson, I.R. Biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma: Twenty years from the bench to the bedside. Cancer J. 2010, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Maini, C.; Manara, M.C.; Benini, S.; Serra, M.; Cerisano, V.; Strammiello, R.; Baldini, N.; Lollini, P.L.; Nanni, P.; et al. Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing’s sarcoma cells. Cancer Gene. Ther. 2002, 9, 296–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Maldegem, A.M.; Bovée, J.V.; Peterse, E.F.; Hogendoorn, P.C.; Gelderblom, H. Ewing sarcoma: The clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur. J. Cancer 2016, 53, 171–180. [Google Scholar] [CrossRef]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2014, 32, 2059–2066. [Google Scholar] [CrossRef]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef]
- DuBois, S.; Bender, J.G.; Buxton, A.; Laack, N.; Randall, L.; Chen, H.; Seibel, N.; Terezakis, S.; Hill-Kayser, C.; Hayes-Jordan, A.; et al. Randomized phase 3 trial of ganitumab added to interval compressed chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma: A report from the Children’s Oncology Group (COG). In Proceedings of the Connective Tissue Oncology Society Annual Meeting, Tokyo, Japan, 13–16 November 2019. [Google Scholar]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Demetri, G.D.; Chawla, S.P.; Ray-Coquard, I.; Le Cesne, A.; Staddon, A.P.; Milhem, M.M.; Penel, N.; Riedel, R.F.; Bui-Nguyen, B.; Cranmer, L.D.; et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J. Clin. Oncol. 2013, 31, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.; Folpe, A.L.; Takayama, T.K.; Yeung, R.S. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum. Pathol. 2007, 38, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, R.; Jones, R.L.; Blay, J.Y.; Le Cesne, A.; Provenzano, S.; Antoniou, G.; Mir, O.; Fucà, G.; Fumagalli, E.; Bertulli, R.; et al. Role of chemotherapy, VEGFR inhibitors, and mTOR inhibitors in advanced perivascular epithelioid cell tumors (PEComas). Clin. Cancer Res. 2019, 25, 5295–5300. [Google Scholar] [CrossRef] [PubMed]
- Binh, M.B.; Sastre-Garau, X.; Guillou, L.; de Pinieux, G.; Terrier, P.; Lagacé, R.; Aurias, A.; Hostein, I.; Coindre, J.M. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: A comparative analysis of 559 soft tissue neoplasms with genetic data. Am. J. Surg. Pathol. 2005, 29, 1340–1347. [Google Scholar] [CrossRef]
- Bill, K.L.; Garnett, J.; Meaux, I.; Ma, X.; Creighton, C.J.; Bolshakov, S.; Barriere, C.; Debussche, L.; Lazar, A.J.; Prudner, B.C.; et al. SAR405838: A novel and potent inhibitor of the MDM2:p53 axis for the treatment of dedifferentiated liposarcoma. Clin. Cancer Res. 2016, 22, 1150–1160. [Google Scholar] [CrossRef]
- Gounder, M.; Bauer, T.M.; Schwartz, G.K.; Weise, A.M.; LoRusso, P.; Kumar, P.; Chen, S.; Mendell, J.; Kochan, J.; Zernovak, O.; et al. Milademetan, an oral MDM2 inhibitor, in well-differentiated/de-differentiated liposarcoma: Results from a phase 1 study in patients with solid tumor and lymphomas. In Proceedings of the Connective Tissue Oncology Society Annual Meeting, Rome, Italy, 14–17 November 2018. [Google Scholar]
- Dickson, M.A.; Tap, W.D.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Rathbone, D.D.; Condy, M.M.; et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [Google Scholar] [CrossRef]
- Dickson, M.A.; D’Angelo, S.; Gounder, M.; Keohan, M.L.; Kelly, C.M.; Chi, P.; Antonescu, C.; Landa, J.; Qin, L.X.; Koff, A.; et al. Phase 2 study of the CDK4 inhibitor abemaciclib in differentiated liposarcoma. In Proceedings of the Connective Tissue Oncology Society Annual Meeting, Rome, Italy, 14–17 November 2018. [Google Scholar]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Nakayama, R.; Zhang, Y.X.; Czaplinski, J.T.; Anatone, A.J.; Sicinska, E.T.; Fletcher, J.A.; Demetri, G.D.; Wagner, A.J. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget 2016, 7, 16581–16592. [Google Scholar] [CrossRef]
- Gounder, M.M.; Zer, A.; Tap, W.D.; Salah, S.; Dickson, M.A.; Gupta, A.A.; Keohan, M.L.; Loong, H.H.; D’Angelo, S.P.; Baker, S.; et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J. Clin. Oncol. 2016, 34, 3166–3174. [Google Scholar] [CrossRef]
- Gounder, M.M.; Somaiah, N.; Attia, S.; Chawla, S.P.; Villalobos, V.M.; Chmielowski, B.; Burgess, M.A.; Schwartz, G.K.; Riedel, R.F.; von Mehren, M.; et al. Phase 2 results of selinexor in advanced de-differentiated (DDLS) liposarcoma (SEAL) study: A phase 2/3, randomized, double blind, placebo controlled cross-over study. J. Clin. Oncol. 2018, 36, 11512. [Google Scholar] [CrossRef]
- Boxberg, M.; Steiger, K.; Lenze, U.; Rechl, H.; von Eisenhart-Rothe, R.; Wörtler, K.; Weichert, W.; Langer, R.; Specht, K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue—Prognostic implications and rationale for immunotherapy. Oncoimmunology 2017, 7, e1389366. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, W.; Wan, P.; Jiang, X.; Chen, J.; Zheng, Y.; Li, W.; Tan, J.; Zhang, S. Clinicopathological and prognostic significance of PD-L1 expression in sarcoma: A systematic review and meta-analysis. Med. Baltim. 2018, 97, e11004. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Middha, S.; Zhang, L.; Nafa, K.; Jayakumaran, G.; Wong, D.; Kim, H.R.; Sadowska, J.; Berger, M.F.; Delair, D.F.; Shia, J.; et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Coyne, G.O.; Sharon, E.; Moore, N.; Meehan, R.; Takebe, N.; Juwara, L.; Rubinstein, L.; Read, W.; Riedel, R.F.; Merriam, P.; et al. Phase II study of atezolizumab in patients with alveolar soft part sarcoma. In Proceedings of the Connective Tissue Oncology Society Annual Meeting, Rome, Italy, 14–17 November 2018. [Google Scholar]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Maki, R.G. Diagnosis, prognosis, and treatment of alveolar soft-part sarcoma: A review. JAMA Oncol. 2019, 5, 254–260. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Gupta, A.; Lipson, D.; Otto, G.; Brennan, T.; Chung, C.T.; Borinstein, S.C.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014, 4, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef]
- Solomon, J.P.; Linkov, I.; Rosado, A.; Mullaney, K.; Rosen, E.Y.; Frosina, D.; Jungbluth, A.A.; Zehir, A.; Benayed, R.; Drilon, A.; et al. NTRK fusion detection across multiple assays and 33,997 cases: Diagnostic implications and pitfalls. Mod. Pathol. 2019. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.M.; Hornick, J.L. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology 2018, 73, 634–644. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.; Dickson, B.C.; Swanson, D.; Zhang, L.; Sung, Y.S.; Huang, H.Y.; Fletcher, C.D.; Antonescu, C.R. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer 2019, 58, 739–746. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Sambri, A.; Bianchi, G.; Cevolani, L.; Donati, D.; Abudu, A. Can radical margins improve prognosis in primary and localized epithelioid sarcoma of the extremities? J. Surg. Oncol. 2018, 117, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Sambri, A.; Pedrini, E.; Pazzaglia, L.; Sangiorgi, L.; Ruengwanichayakun, P.; Donati, D.; Benassi, M.S.; Righi, A. Histological and molecular features of solitary fibrous tumor of the extremities: Clinical correlation. Virchows Arch. 2019. [Google Scholar] [CrossRef] [PubMed]
- Prlestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haldarl, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed]
- Sunami, K.; Ichikawa, H.; Kubo, T.; Kato, M.; Fujiwara, Y.; Shimomura, A.; Koyama, T.; Kakishima, H.; Kitami, M.; Matsushita, H.; et al. Feasibility and utility of a panel testing for 114 cancer-associated genes in a clinical setting: A hospital-based study. Cancer Sci. 2019, 110, 1480–1490. [Google Scholar] [CrossRef]
- Sambri, A.; Bianchi, G.; Cucurnia, I.; Gambarotti, M.; Donati, D.M. Pediatric soft tissue sarcoma of the limbs: Clinical outcome of 97 patients. Eur. J. Orthop. Surg. Traumatol. 2018, 28, 1–7. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Trédan, O.; Wang, Q.; Pissaloux, D.; Cassier, P.; de la Fouchardière, A.; Fayette, J.; Desseigne, F.; Ray-Coquard, I.; de la Fouchardière, C.; Frappaz, D.; et al. ProfiLER investigators. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: Analysis from the ProfiLER trial. Ann. Oncol. 2019, 30, 757–765. [Google Scholar] [CrossRef]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Corao, D.A.; Biegel, J.A.; Coffin, C.M.; Barr, F.G.; Wainwright, L.M.; Ernst, L.M.; Choi, J.K.; Zhang, P.J.; Pawel, B.R. ALK expression in rhabdomyosarcomas: Correlation with histologic subtype and fusion status. Pediatr. Dev. Pathol. 2009, 12, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Stacchiotti, S.; Rutkowski, P.; Blay, J.Y.; Lindner, L.H.; Strauss, S.J.; Anthoney, A.; Duffaud, F.; Richter, S.; et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 ‘CREATE’. Ann. Oncol. 2017, 28, 3000–3008. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.D.; Shankaran, V.; Sullivan, S.D. Basket Cases: How real-world testing for drugs approved based on basket trials might lead to false diagnoses, patient risks, and squandered resources. J. Clin. Oncol. 2019, 36, 3472–3474. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Nepom, G.T.; Lonial, S. Academic, Foundation, and industry collaboration in finding new therapies. N. Engl. J. Med. 2017, 376, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.G.; Haider, A.H.; Landman, A.B.; Raut, C.P. The opportunities and shortcomings of using big data and national databases for sarcoma research. Cancer 2019, 125, 2926–2934. [Google Scholar] [CrossRef]
- Woodcock, J.; LaVange, L.M. Master protocols to study multiple therapies, multiple diseases, or both. N. Engl. J. Med. 2017, 377, 62–70. [Google Scholar] [CrossRef]

{kind=link}
| Trial Name | Antitumor Drug Adding to Doxorubicin a | Median OS (Months) | Hazard Ratio [95%CI] | p-Value | Ref. |
|---|---|---|---|---|---|
| EORTC 62012 | Ifosfamide | 14.3 vs. 12.8 | 0.83 [0.67, 1.03] | 0.076 | [6] |
| PICASSO III | Palifosfamide | 15.9 vs. 16.9 | 1.05 [0.79, 1.39] | 0.74 | [7] |
| SARC021 | Evofosfamide | 18.4 vs. 19.0 | 1.06 [0.88, 1.29] | 0.527 | [8] |
| ANNOUNCE | Olaratumab | 20.4 vs. 19.8 | 1.05 [0.84, 1.30] | 0.69 | [9] |
| Antitumor Drug | Enrollment by Histological Subtype | Ref. | |||
|---|---|---|---|---|---|
| Leiomyo-Sarcoma | Liposarcoma | Synovial Sarcoma | Undifferentiated Pleomorphic Sarcoma | ||
| Pazopanib | 〇 | × | 〇 | 〇 | [22] |
| Trabectedin | 〇 | 〇 | × | × | [23] |
| Eribulin | 〇 | 〇 | × | × | [24] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, K.; Takahashi, S. Precision Medicine in Soft Tissue Sarcoma Treatment. Cancers 2020, 12, 221. https://doi.org/10.3390/cancers12010221
Nakano K, Takahashi S. Precision Medicine in Soft Tissue Sarcoma Treatment. Cancers. 2020; 12(1):221. https://doi.org/10.3390/cancers12010221
Chicago/Turabian StyleNakano, Kenji, and Shunji Takahashi. 2020. "Precision Medicine in Soft Tissue Sarcoma Treatment" Cancers 12, no. 1: 221. https://doi.org/10.3390/cancers12010221
APA StyleNakano, K., & Takahashi, S. (2020). Precision Medicine in Soft Tissue Sarcoma Treatment. Cancers, 12(1), 221. https://doi.org/10.3390/cancers12010221