Single-Cell Analysis of Multiple Steps of Dynamic NF-κB Regulation in Interleukin-1α-Triggered Tumor Cells Using Proximity Ligation Assays


 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
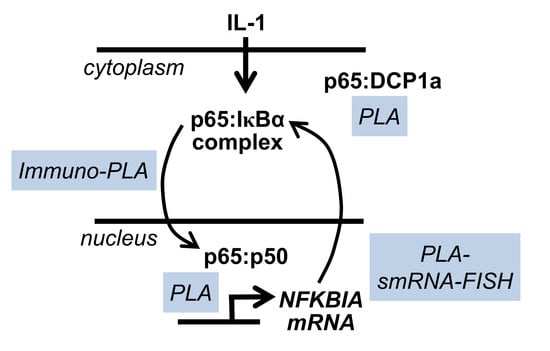
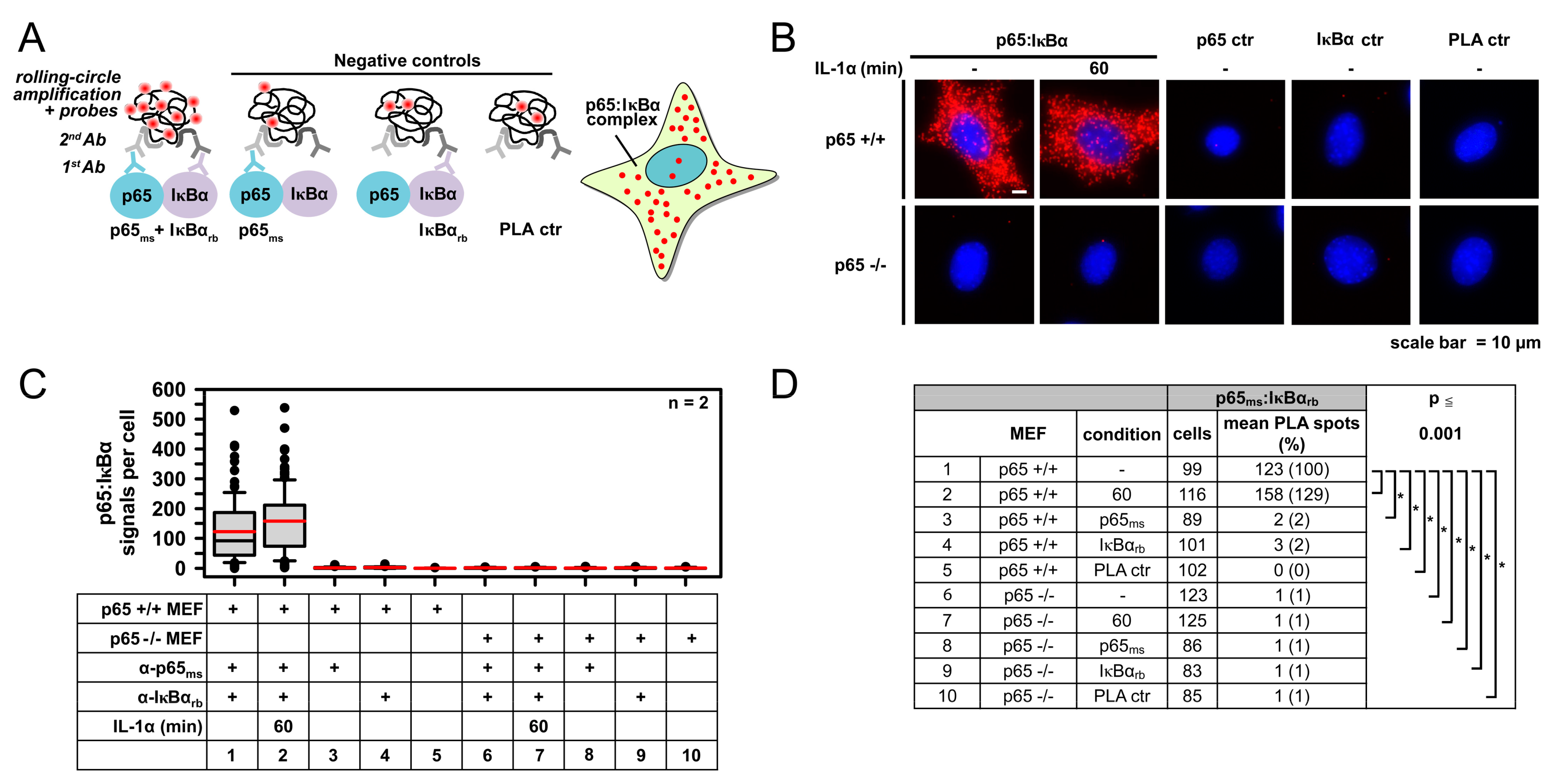
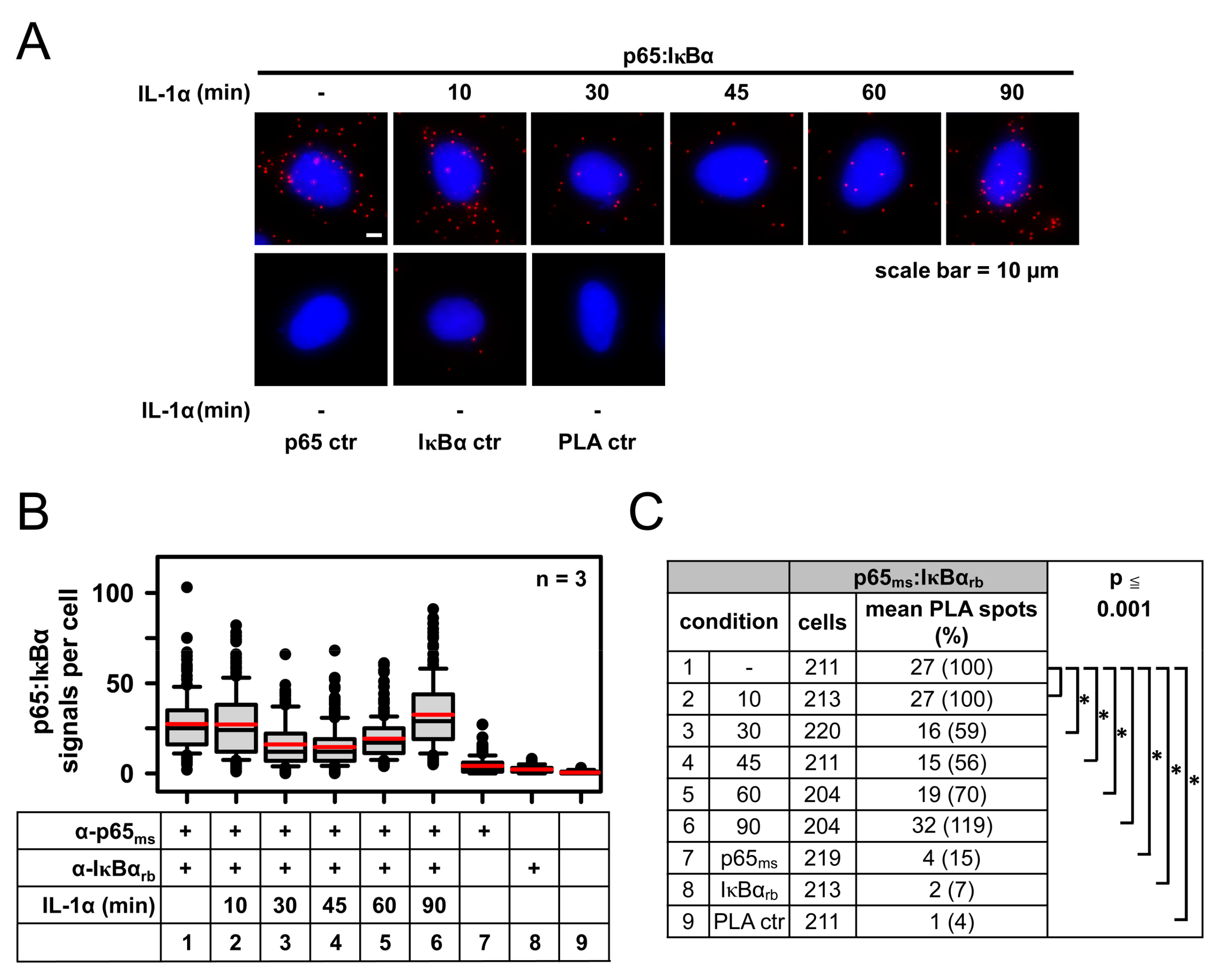
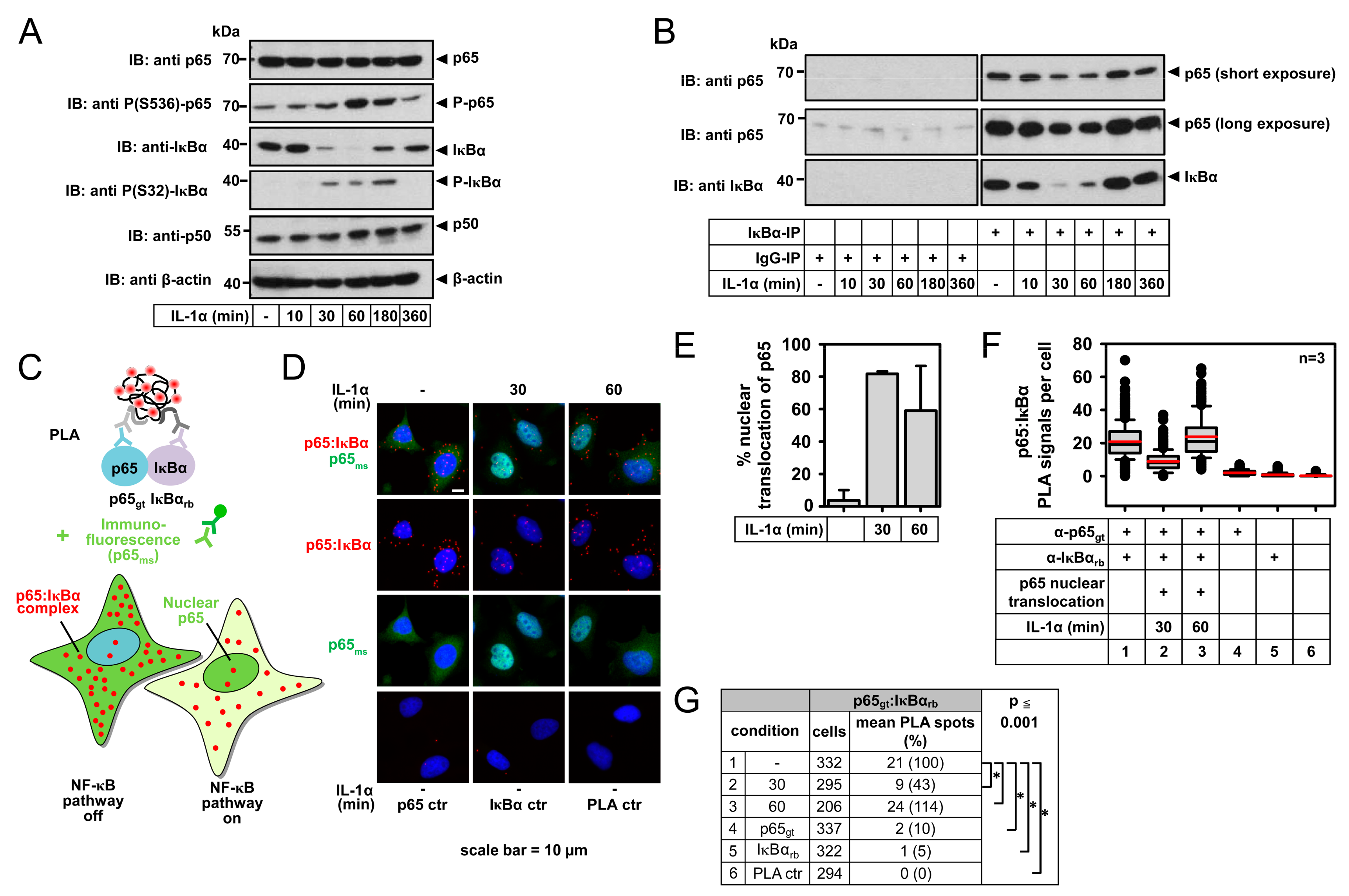
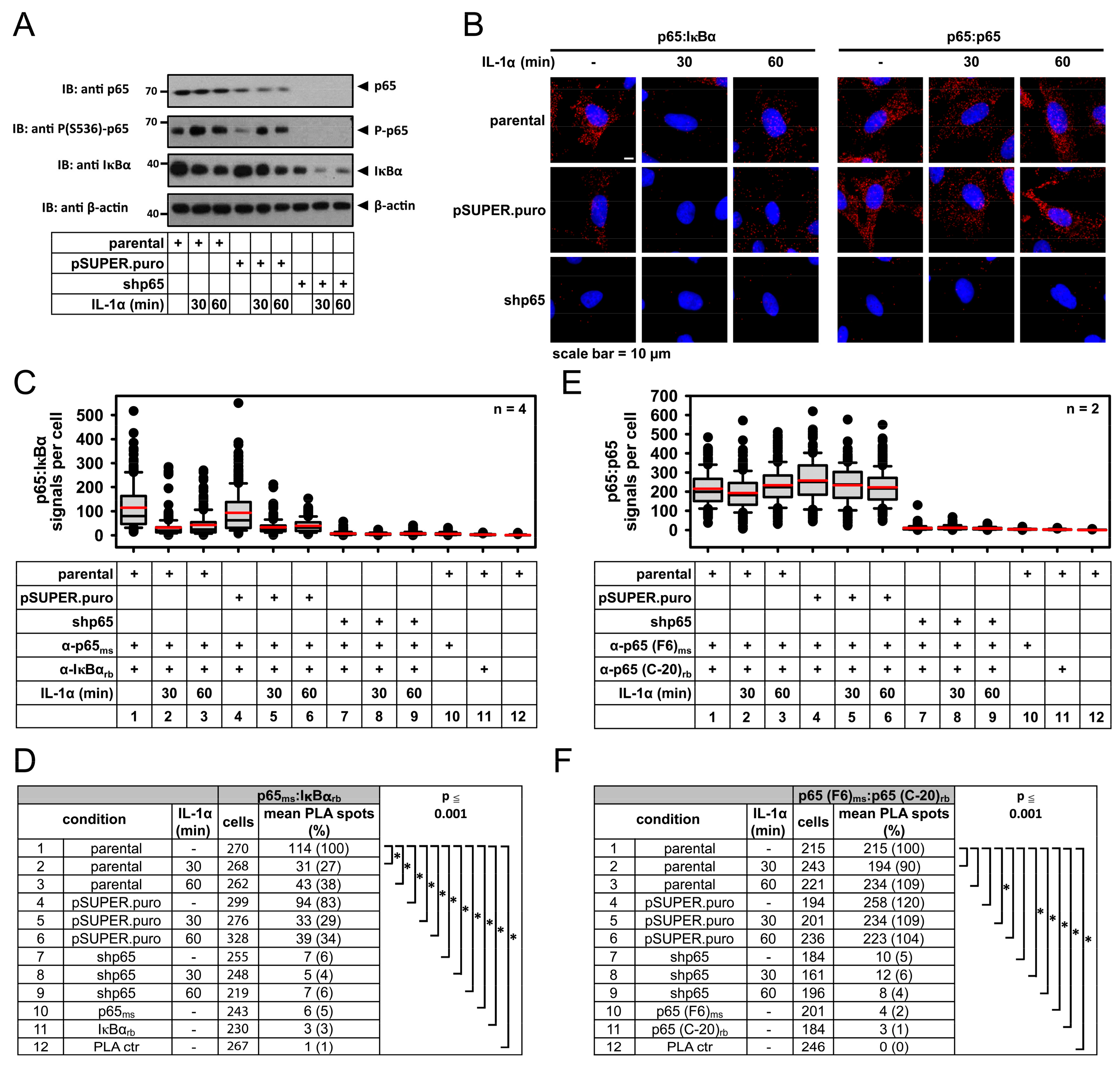
2.1. IL-1α-Dependent Dynamic Regulation of Endogenous p65/IκBα Complexes in Single Cells
2.2. Specificity of PLA in Human Cancer Cells and Its Use to Determine p65 Protein Levels
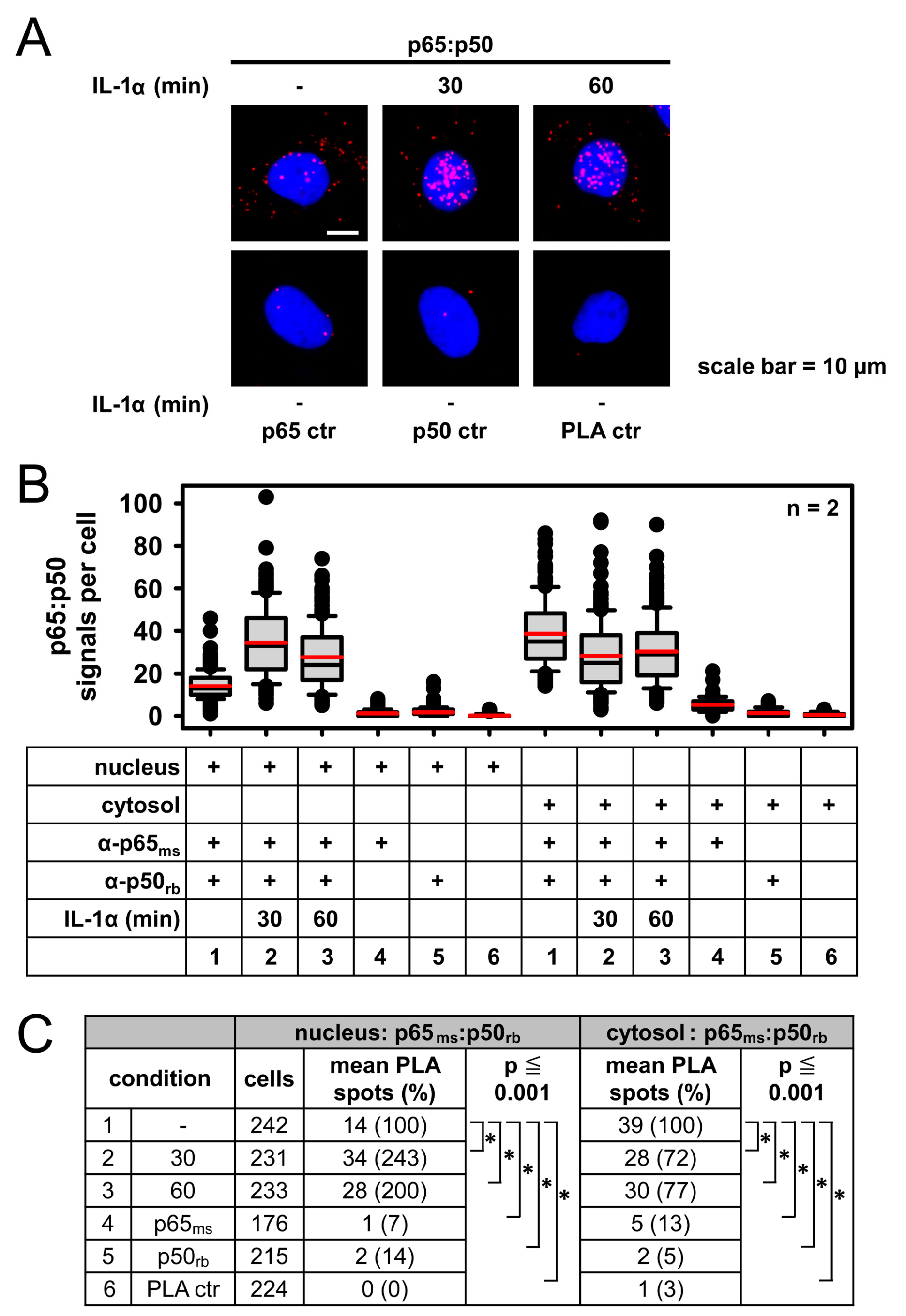
2.3. Detection of IL-1α-Regulated Active Nuclear NF-κB Complexes by PLA
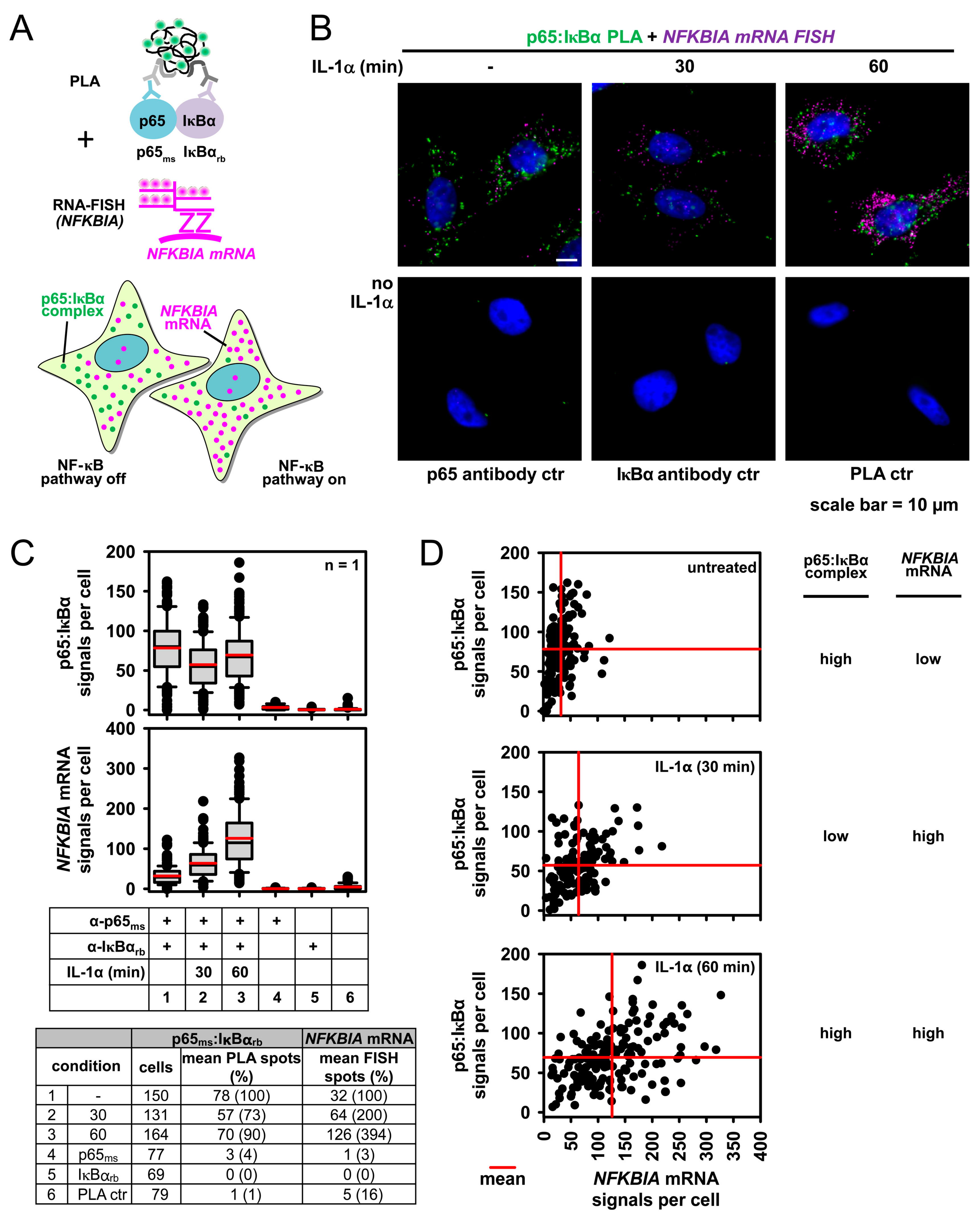
2.4. Combining PLA with Single Cell Gene Expression Analysis Using Single Molecule RNA Fluorescence in Situ Hybridization (smRNA-FISH)
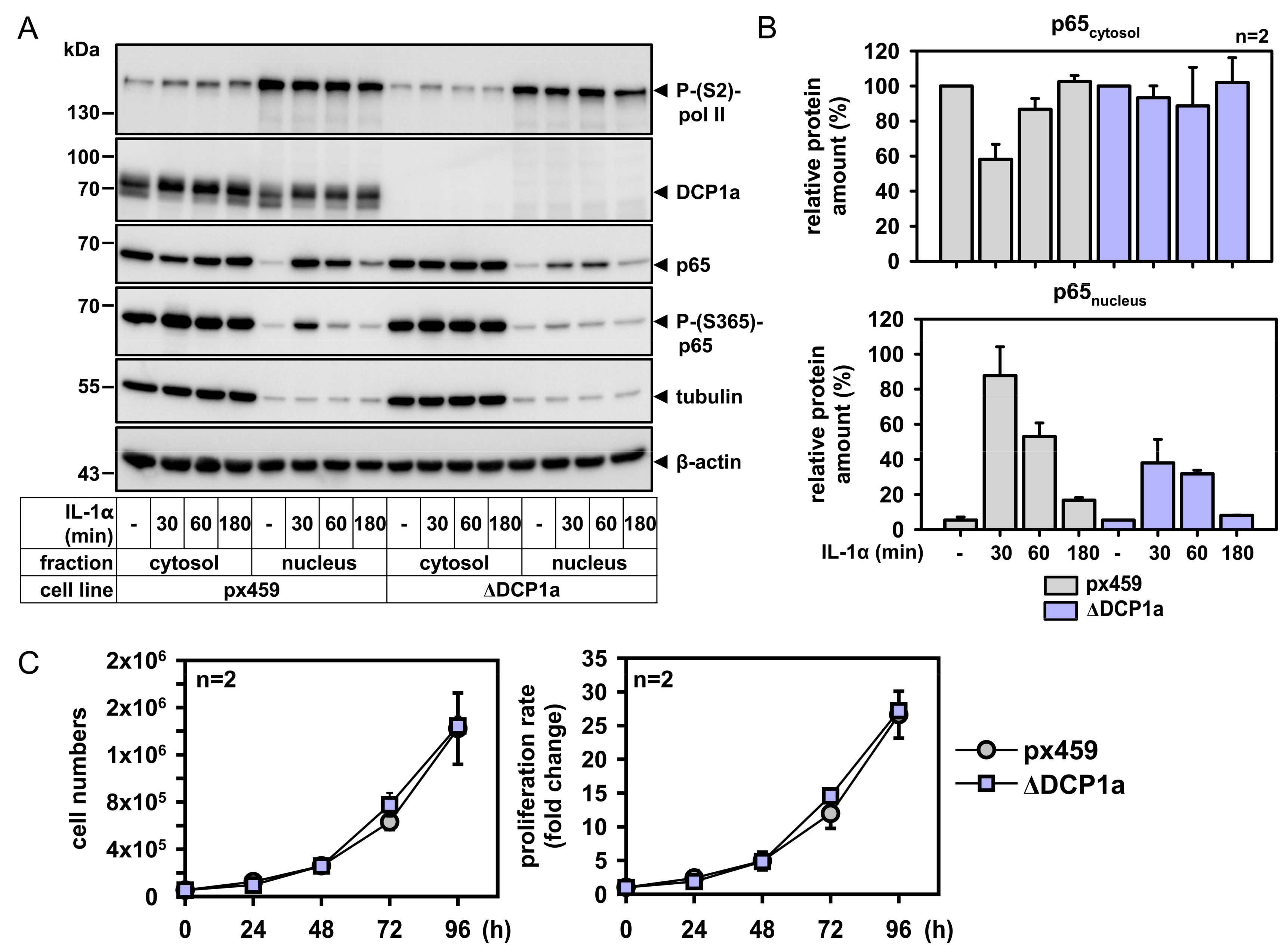
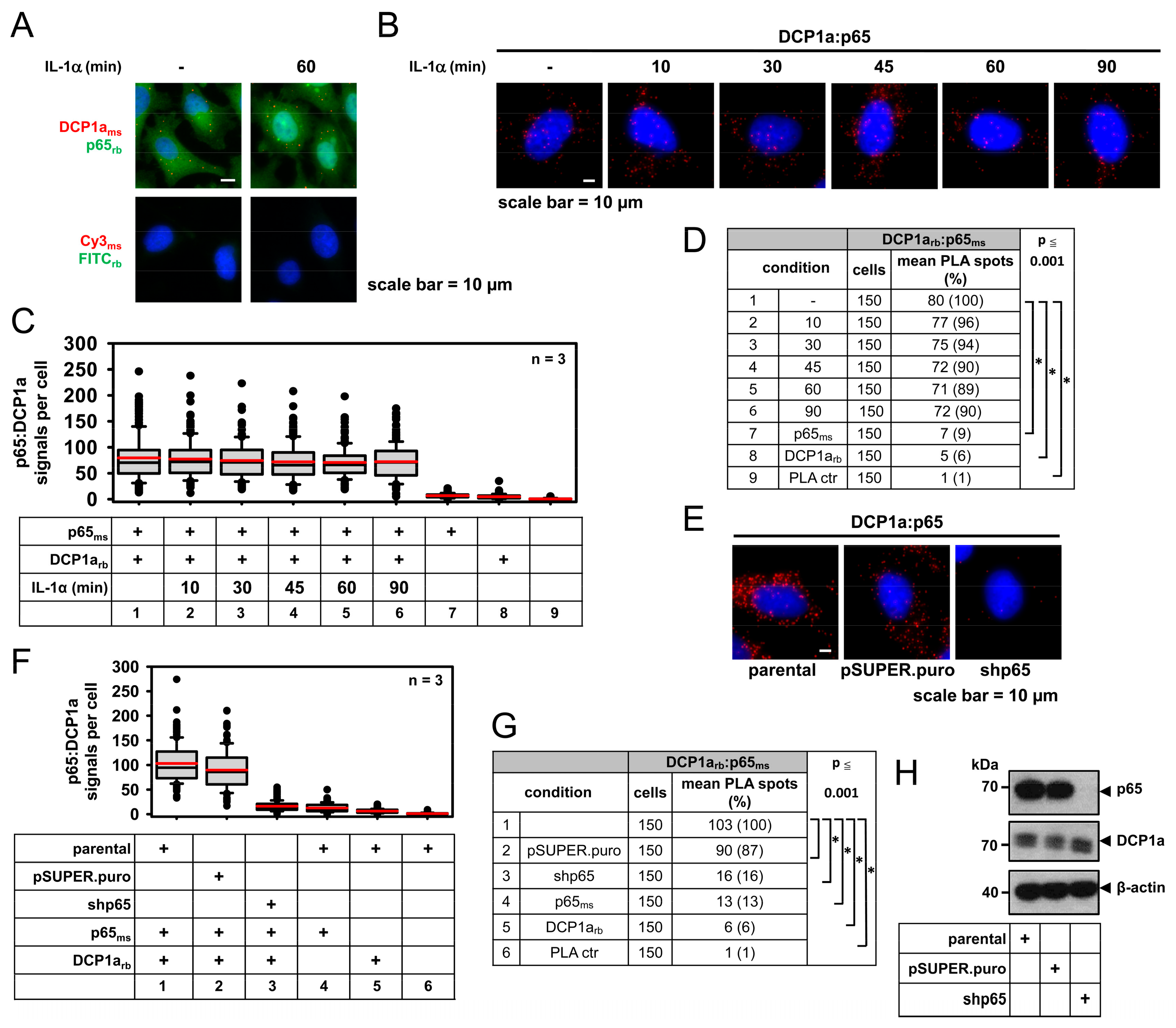
2.5. DCP1a Constitutively Associates with p65 to Indirectly Control Its IL-1α-Triggered Nuclear Import
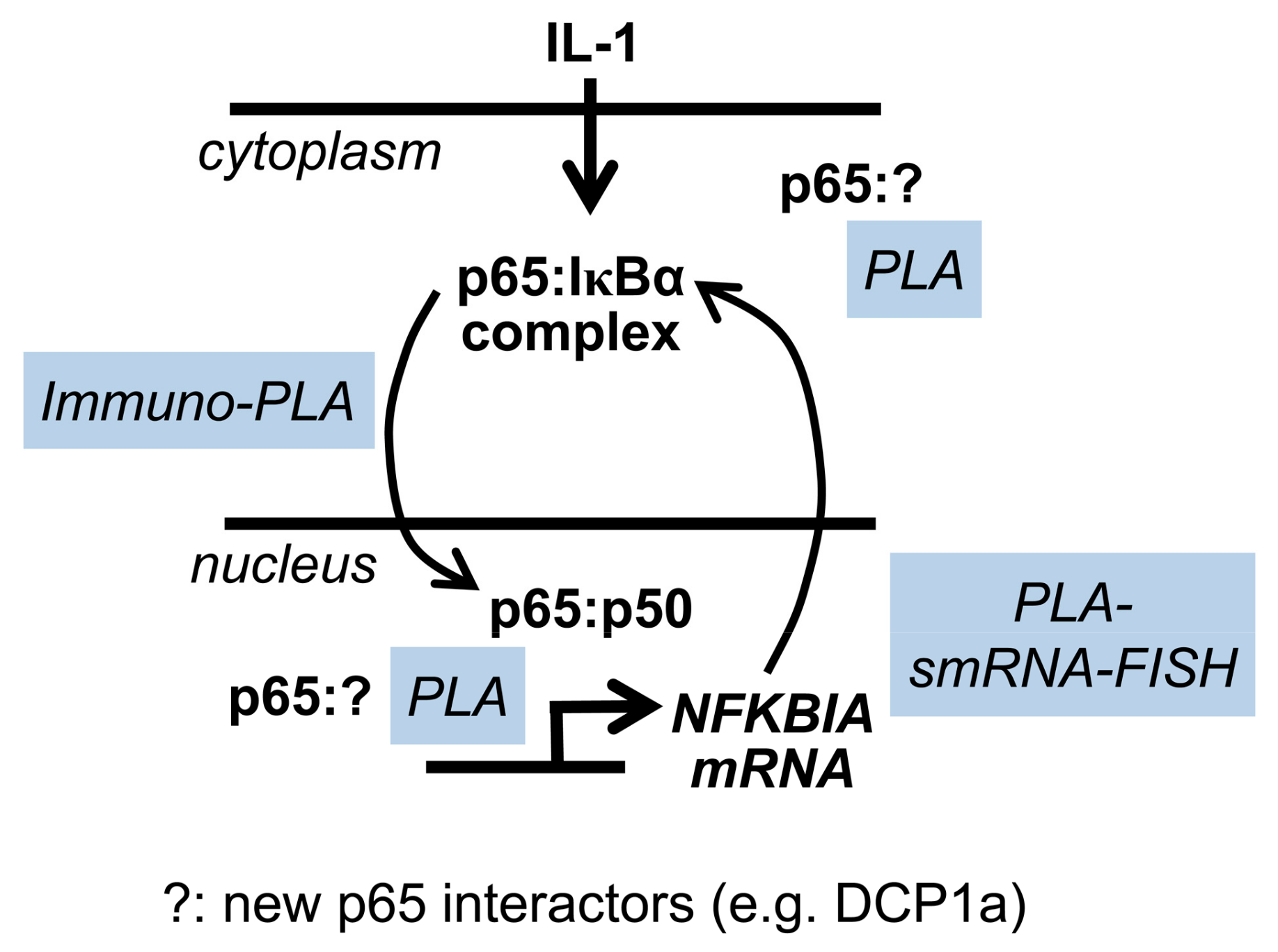
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids, Transient, or Stable Transfections
4.3. (Immuno)-Proximity Ligation Assays
4.4. PLA Coupled to Single Molecule RNA-Fluorescence in Situ Hybridization (PLA-smRNA-FISH)
4.5. Software Used, Quantification, and Statistical Analyses
4.6. Cell Lysis, Western Blotting, and Co-Immunoprecipitation Experiments
4.7. Antibodies and Reagents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular plasticity in cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with nf-kappab as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Riedlinger, T.; Haas, J.; Busch, J.; van de Sluis, B.; Kracht, M.; Schmitz, M.L. The direct and indirect roles of nf-kappab in cancer: Lessons from oncogenic fusion proteins and knock-in mice. Biomedicines 2018, 6, 36. [Google Scholar] [CrossRef]
- Worzfeld, T.; Pogge von Strandmann, E.; Huber, M.; Adhikary, T.; Wagner, U.; Reinartz, S.; Muller, R. The unique molecular and cellular microenvironment of ovarian cancer. Front. Oncol. 2017, 7, 24. [Google Scholar] [CrossRef]
- Ren, X.; Kang, B.; Zhang, Z. Understanding tumor ecosystems by single-cell sequencing: Promises and limitations. Genome Biol. 2018, 19, 211. [Google Scholar] [CrossRef]
- Packer, J.; Trapnell, C. Single-cell multi-omics: An engine for new quantitative models of gene regulation. Trends Genet. 2018, 34, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Doerr, A. Single-cell proteomics. Nat. Methods 2019, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Weibrecht, I.; Lundin, E.; Kiflemariam, S.; Mignardi, M.; Grundberg, I.; Larsson, C.; Koos, B.; Nilsson, M.; Soderberg, O. In situ detection of individual mrna molecules and protein complexes or post-translational modifications using padlock probes combined with the in situ proximity ligation assay. Nat. Protoc. 2013, 8, 355–372. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gustafsdottir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Petruk, S.; Fenstermaker, T.K.; Black, K.L.; Brock, H.W.; Mazo, A. Detection of rna-DNA association by a proximity ligation-based method. Sci. Rep. 2016, 6, 27313. [Google Scholar] [CrossRef]
- Roussis, I.M.; Guille, M.; Myers, F.A.; Scarlett, G.P. Rna whole-mount in situ hybridisation proximity ligation assay (rish-pla), an assay for detecting rna-protein complexes in intact cells. PLoS ONE 2016, 11, e0147967. [Google Scholar] [CrossRef]
- Smith, M.A.; Hall, R.; Fisher, K.; Haake, S.M.; Khalil, F.; Schabath, M.B.; Vuaroqueaux, V.; Fiebig, H.H.; Altiok, S.; Chen, Y.A.; et al. Annotation of human cancers with egfr signaling-associated protein complexes using proximity ligation assays. Sci. Signal. 2015, 8, ra4. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. Nf-kappab, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of nf-kappab subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The nf-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the nfkappab system. Wiley. Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth ikappab protein within the nf-kappab signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of nf-kappab proteins. Cold Spring Harb. Perspect. Biol. 2009, 1, a000067. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Nf-kappab, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The ikappab kinase complex (ikk) contains two kinase subunits, ikkalpha and ikkbeta, necessary for ikappab phosphorylation and nf-kappab activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. Ikk-1 and ikk-2: Cytokine-activated ikappab kinases essential for nf-kappab activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the ikappabalpha-ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [CrossRef]
- Malek, S.; Chen, Y.; Huxford, T.; Ghosh, G. Ikappabbeta, but not ikappabalpha, functions as a classical cytoplasmic inhibitor of nf-kappab dimers by masking both nf-kappab nuclear localization sequences in resting cells. J. Biol. Chem. 2001, 276, 45225–45235. [Google Scholar] [CrossRef]
- Schüller, R.; Forné, I.; Straub, T.; Schreieck, A.; Texier, Y.; Shah, N.; Decker, T.M.; Cramer, P.; Imhof, A.; Eick, D. Heptad-specific phosphorylation of rna polymerase ii ctd. Mol. Cell 2016, 61, 305–314. [Google Scholar] [CrossRef]
- Renner, F.; Schmitz, M.L. Autoregulatory feedback loops terminating the nf-kappab response. Trends Biochem. Sci. 2009, 34, 128–135. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Weber, A.; Roxlau, T.; Gaestel, M.; Kracht, M. Signal integration, crosstalk mechanisms and networks in the function of inflammatory cytokines. Biochim. Biophys. Acta 2011, 1813, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Arenzana-Seisdedos, F.; Turpin, P.; Rodriguez, M.; Thomas, D.; Hay, R.T.; Virelizier, J.L.; Dargemont, C. Nuclear localization of i kappa b alpha promotes active transport of nf-kappa b from the nucleus to the cytoplasm. J. Cell Sci. 1997, 110, 369–378. [Google Scholar] [PubMed]
- Griffin, B.D.; Moynagh, P.N. In vivo binding of nf-kappab to the ikappabbeta promoter is insufficient for transcriptional activation. Biochem. J. 2006, 400, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Holtmann, H.; Gaestel, M. The role of mammalian mapk signaling in regulation of cytokine mrna stability and translation. J. Interferon Cytokine Res. 2014, 34, 220–232. [Google Scholar] [CrossRef]
- Tiedje, C.; Diaz-Munoz, M.D.; Trulley, P.; Ahlfors, H.; Laass, K.; Blackshear, P.J.; Turner, M.; Gaestel, M. The rna-binding protein ttp is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. 2016, 44, 7418–7440. [Google Scholar] [CrossRef] [PubMed]
- Mikuda, N.; Kolesnichenko, M.; Beaudette, P.; Popp, O.; Uyar, B.; Sun, W.; Tufan, A.B.; Perder, B.; Akalin, A.; Chen, W.; et al. The ikappab kinase complex is a regulator of mrna stability. EMBO J. 2018, 37, e98658. [Google Scholar] [CrossRef]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The stress granule transcriptome reveals principles of mrna accumulation in stress granules. Mol. Cell 2017, 68, 808–820. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-bodies: Composition, properties, and functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Standart, N.; Weil, D. P-bodies: Cytosolic droplets for coordinated mrna storage. Trends. Genet. 2018, 34, 612–626. [Google Scholar]
- Hancock, R. Internal organisation of the nucleus: Assembly of compartments by macromolecular crowding and the nuclear matrix model. Biol. Cell 2004, 96, 595–601. [Google Scholar] [CrossRef]
- Jurida, L.; Soelch, J.; Bartkuhn, M.; Handschick, K.; Muller, H.; Newel, D.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Bhuju, S.; et al. The activation of il-1-induced enhancers depends on tak1 kinase activity and nf-kappab p65. Cell Rep. 2015, 10, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, T.; Liefke, R.; Meier-Soelch, J.; Jurida, L.; Nist, A.; Stiewe, T.; Kracht, M.; Schmitz, M.L. Nf-kappab p65 dimerization and DNA-binding is important for inflammatory gene expression. FASEB J. 2019, 33, 4188–4202. [Google Scholar] [CrossRef] [PubMed]
- Rzeczkowski, K.; Beuerlein, K.; Muller, H.; Dittrich-Breiholz, O.; Schneider, H.; Kettner-Buhrow, D.; Holtmann, H.; Kracht, M. C-jun n-terminal kinase phosphorylates dcp1a to control formation of p bodies. J. Cell Biol. 2011, 194, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, A.C.; Perkins, N.D. Nf-kappab and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; Landegren, U. Close encounters—Probing proximal proteins in live or fixed cells. Trends. Biochem. Sci. 2017, 42, 504–515. [Google Scholar] [CrossRef]
- Chang, S.T.; Zahn, J.M.; Horecka, J.; Kunz, P.L.; Ford, J.M.; Fisher, G.A.; Le, Q.T.; Chang, D.T.; Ji, H.; Koong, A.C. Identification of a biomarker panel using a multiplex proximity ligation assay improves accuracy of pancreatic cancer diagnosis. J. Transl. Med. 2009, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Debaize, L.; Jakobczyk, H.; Rio, A.G.; Gandemer, V.; Troadec, M.B. Optimization of proximity ligation assay (pla) for detection of protein interactions and fusion proteins in non-adherent cells: Application to pre-b lymphocytes. Mol. Cytogenet. 2017, 10, 27. [Google Scholar] [CrossRef]
- Sable, R.; Jambunathan, N.; Singh, S.; Pallerla, S.; Kousoulas, K.G.; Jois, S. Proximity ligation assay to study protein-protein interactions of proteins on two different cells. Biotechniques 2018, 65, 149–157. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hiramatsu, H.; Nakamura, S.; Kobayashi, K.; Haraguchi, T.; Iba, H. Tumor suppression via inhibition of swi/snf complex-dependent nf-kappab activation. Sci. Rep. 2017, 7, 11772. [Google Scholar] [CrossRef]
- Klenke, C.; Widera, D.; Engelen, T.; Muller, J.; Noll, T.; Niehaus, K.; Schmitz, M.L.; Kaltschmidt, B.; Kaltschmidt, C. Hsc70 is a novel interactor of nf-kappab p65 in living hippocampal neurons. PLoS ONE 2013, 8, e65280. [Google Scholar] [CrossRef]
- Zabel, U.; Schreck, R.; Baeuerle, P.A. DNA binding of purified transcription factor nf-kappa b. Affinity, specificity, zn2+ dependence, and differential half-site recognition. J. Biol. Chem. 1991, 266, 252–260. [Google Scholar] [PubMed]
- Tenekeci, U.; Poppe, M.; Beuerlein, K.; Buro, C.; Muller, H.; Weiser, H.; Kettner-Buhrow, D.; Porada, K.; Newel, D.; Xu, M.; et al. K63-ubiquitylation and traf6 pathways regulate mammalian p-body formation and mrna decapping. Mol. Cell 2016, 62, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the ikappab kinase complex by traf6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Haimovich, G.; Medina, D.A.; Causse, S.Z.; Garber, M.; Millan-Zambrano, G.; Barkai, O.; Chavez, S.; Perez-Ortin, J.E.; Darzacq, X.; Choder, M. Gene expression is circular: Factors for mrna degradation also foster mrna synthesis. Cell 2013, 153, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Ramirez, M.J.; Minguillon, J.; Quiles, P.; Ruiz de Garibay, G.; Aza-Carmona, M.; Bogliolo, M.; Pujol, R.; Prados-Carvajal, R.; Fernandez, J.; et al. Decapping protein edc4 regulates DNA repair and phenocopies brca1. Nat. Commun. 2018, 9, 967. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.T.; Bercovich, N.; Loh, B.; Jonas, S.; Izaurralde, E. The activation of the decapping enzyme dcp2 by dcp1 occurs on the edc4 scaffold and involves a conserved loop in dcp1. Nucleic Acids Res. 2014, 42, 5217–5233. [Google Scholar] [CrossRef] [PubMed]
- Handschick, K.; Beuerlein, K.; Jurida, L.; Bartkuhn, M.; Muller, H.; Soelch, J.; Weber, A.; Dittrich-Breiholz, O.; Schneider, H.; Scharfe, M.; et al. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for nf-kappab-dependent gene expression. Mol. Cell 2014, 53, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in il-1-signaling involves deacetylation of p65 nf-kappab. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef]
- Li, H.; Wittwer, T.; Weber, A.; Schneider, H.; Moreno, R.; Maine, G.N.; Kracht, M.; Schmitz, M.L.; Burstein, E. Regulation of nf-kappab activity by competition between rela acetylation and ubiquitination. Oncogene 2012, 31, 611–623. [Google Scholar] [CrossRef]
- Hoffmann, E.; Thiefes, A.; Buhrow, D.; Dittrich-Breiholz, O.; Schneider, H.; Resch, K.; Kracht, M. Mek1-dependent delayed expression of fos-related antigen-1 counteracts c-fos and p65 nf-kappab-mediated interleukin-8 transcription in response to cytokines or growth factors. J. Biol. Chem. 2005, 280, 9706–9718. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayr-Buro, C.; Schlereth, E.; Beuerlein, K.; Tenekeci, U.; Meier-Soelch, J.; Schmitz, M.L.; Kracht, M. Single-Cell Analysis of Multiple Steps of Dynamic NF-κB Regulation in Interleukin-1α-Triggered Tumor Cells Using Proximity Ligation Assays. Cancers 2019, 11, 1199. https://doi.org/10.3390/cancers11081199
Mayr-Buro C, Schlereth E, Beuerlein K, Tenekeci U, Meier-Soelch J, Schmitz ML, Kracht M. Single-Cell Analysis of Multiple Steps of Dynamic NF-κB Regulation in Interleukin-1α-Triggered Tumor Cells Using Proximity Ligation Assays. Cancers. 2019; 11(8):1199. https://doi.org/10.3390/cancers11081199
Chicago/Turabian StyleMayr-Buro, Christin, Eva Schlereth, Knut Beuerlein, Ulas Tenekeci, Johanna Meier-Soelch, M. Lienhard Schmitz, and Michael Kracht. 2019. "Single-Cell Analysis of Multiple Steps of Dynamic NF-κB Regulation in Interleukin-1α-Triggered Tumor Cells Using Proximity Ligation Assays" Cancers 11, no. 8: 1199. https://doi.org/10.3390/cancers11081199
APA StyleMayr-Buro, C., Schlereth, E., Beuerlein, K., Tenekeci, U., Meier-Soelch, J., Schmitz, M. L., & Kracht, M. (2019). Single-Cell Analysis of Multiple Steps of Dynamic NF-κB Regulation in Interleukin-1α-Triggered Tumor Cells Using Proximity Ligation Assays. Cancers, 11(8), 1199. https://doi.org/10.3390/cancers11081199