Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance

 , and
, and 
Abstract
1. Introduction
2. Results
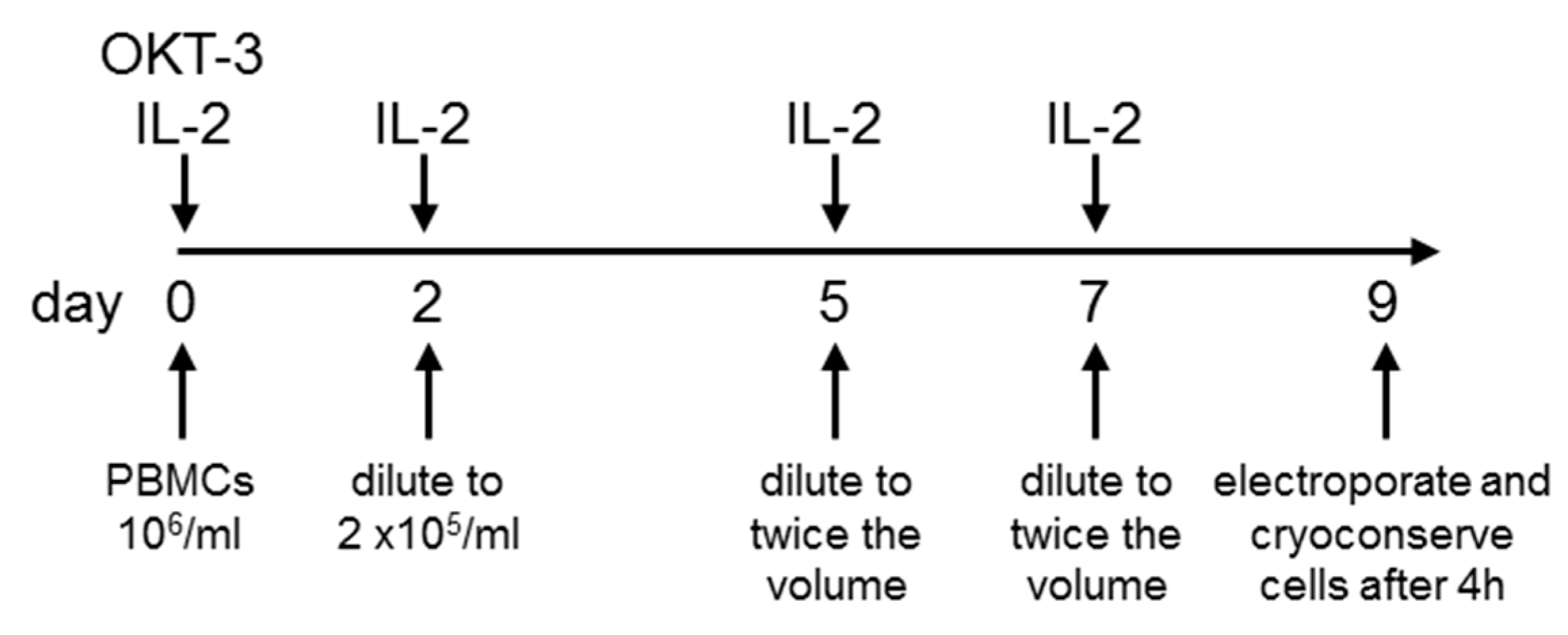
2.1. Sufficient Numbers of CAR-Transfected T Cells Are Generated from PBMCs Originating from Leukaphereses
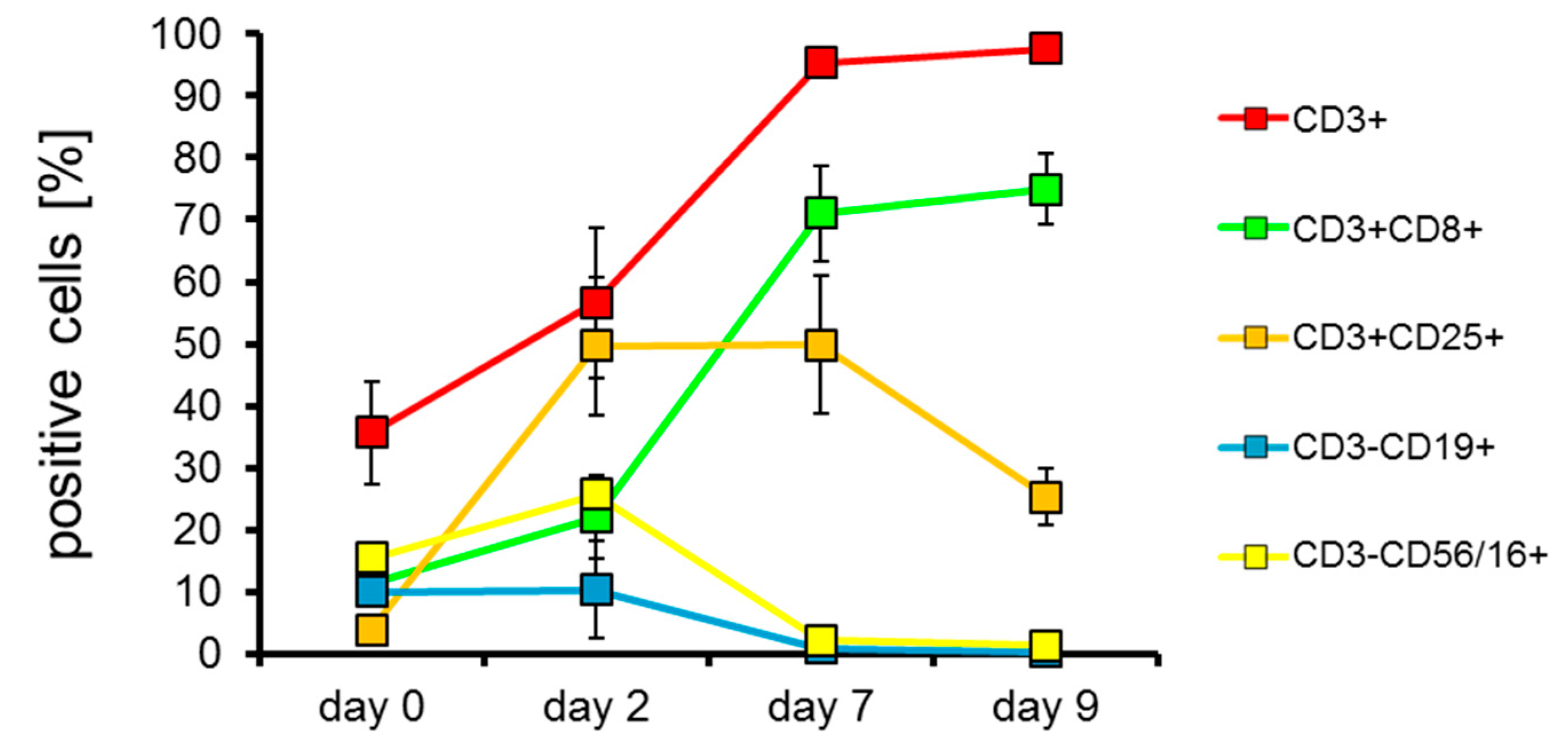
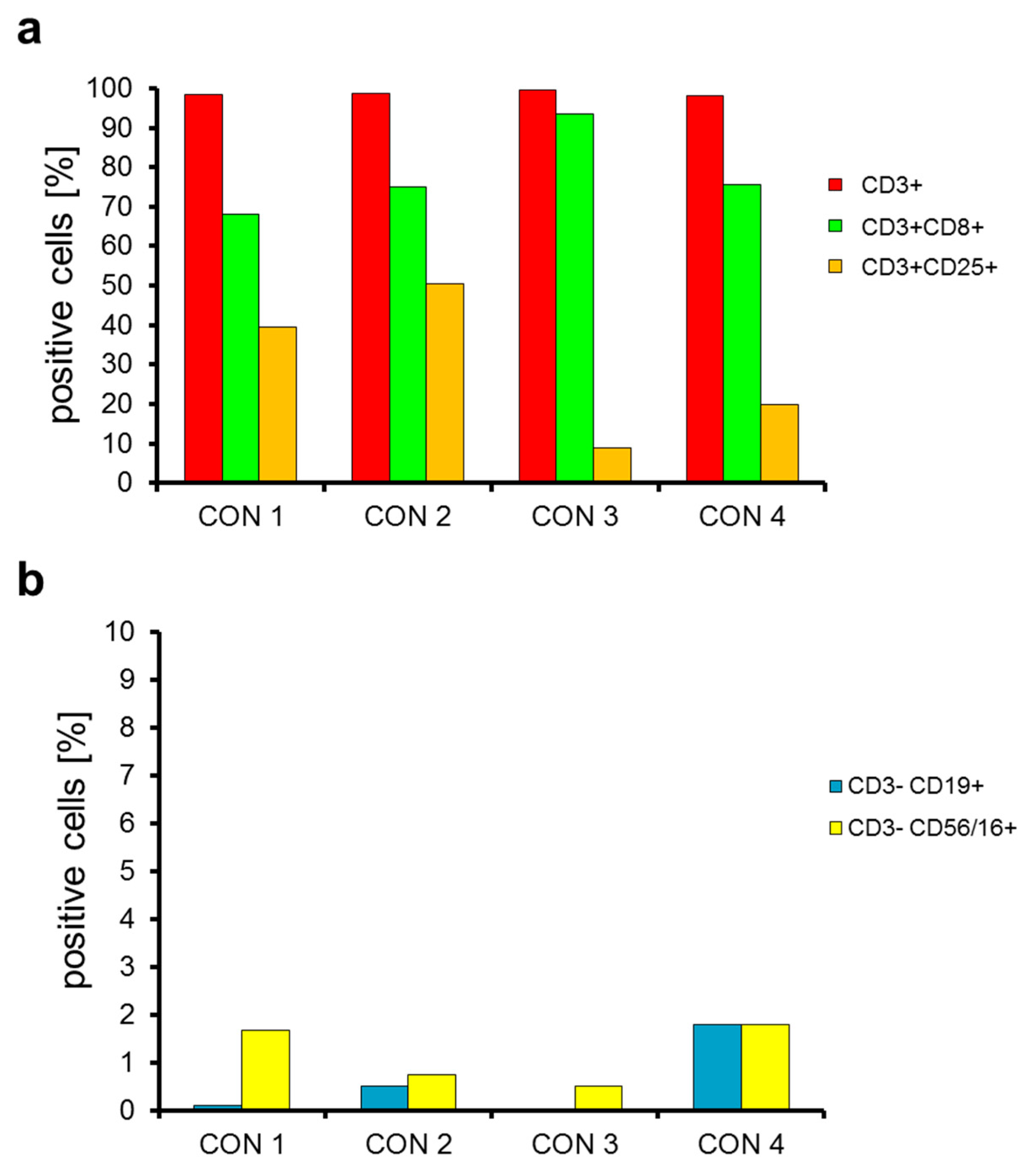
2.2. T Cells Are Preferentially Expanded over 9 Days
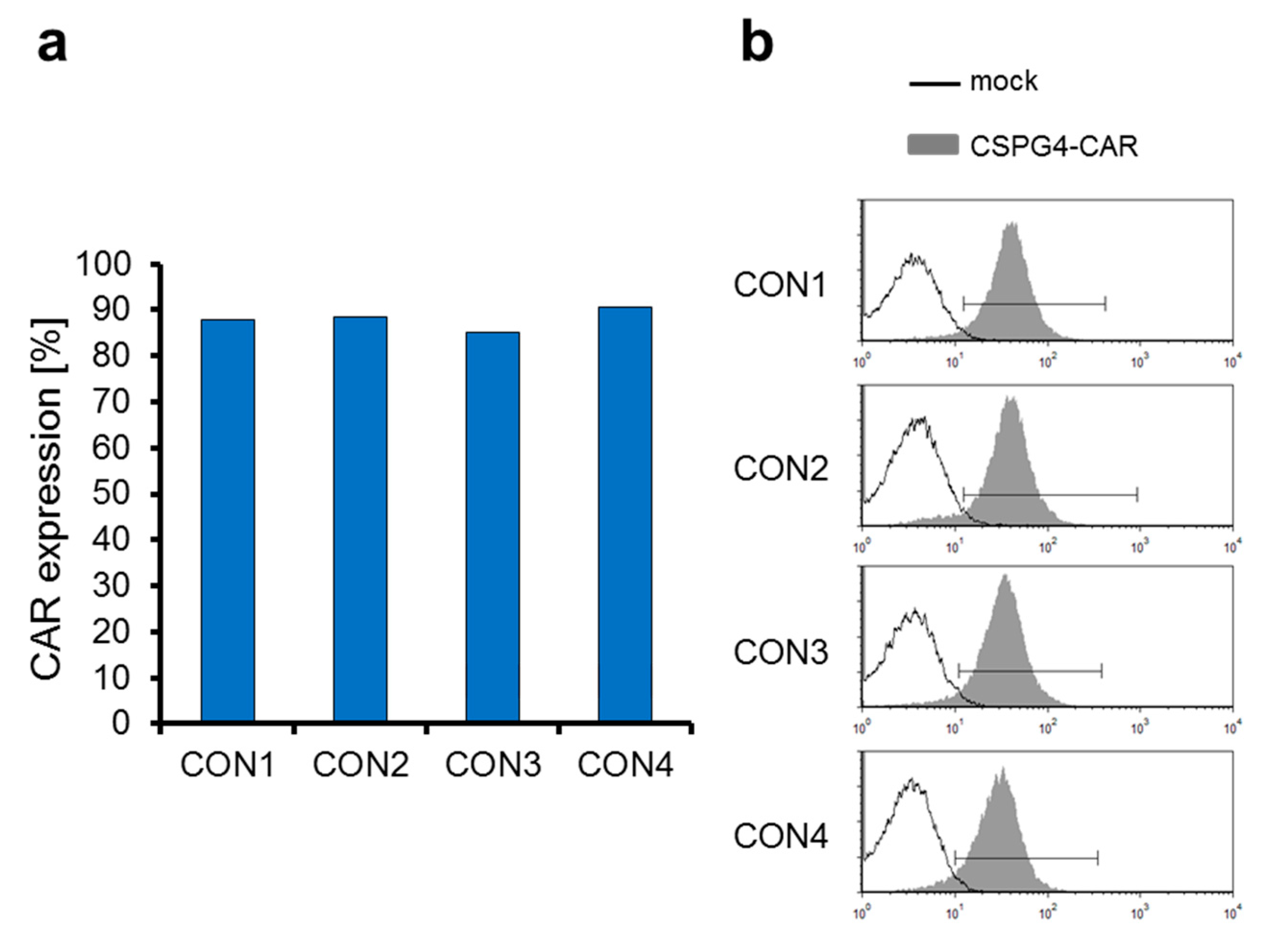
2.3. The CSPG4-Specific CAR Is Expressed Very Efficiently on T Cells Electroporated with CAR-Encoding mRNA
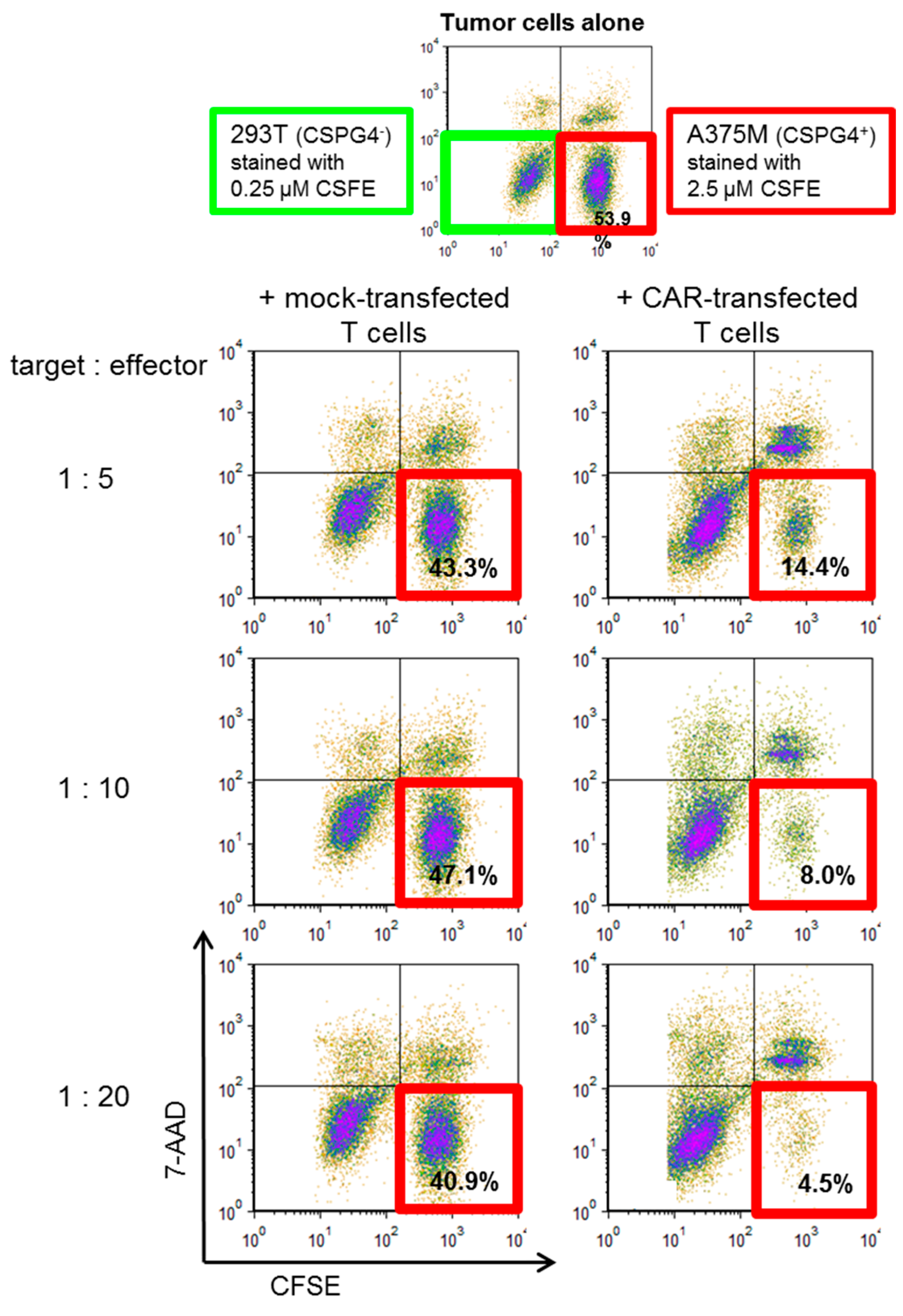
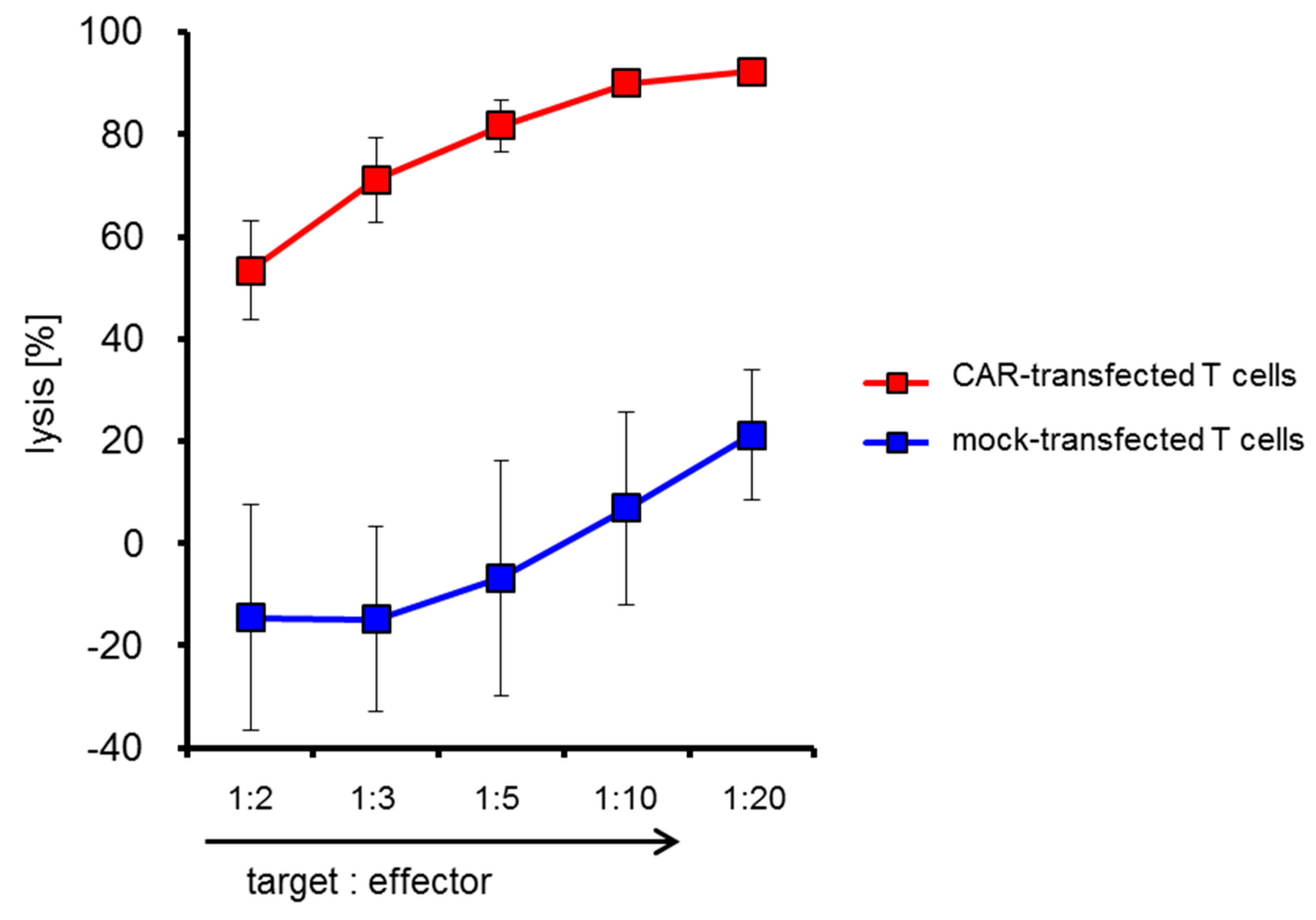
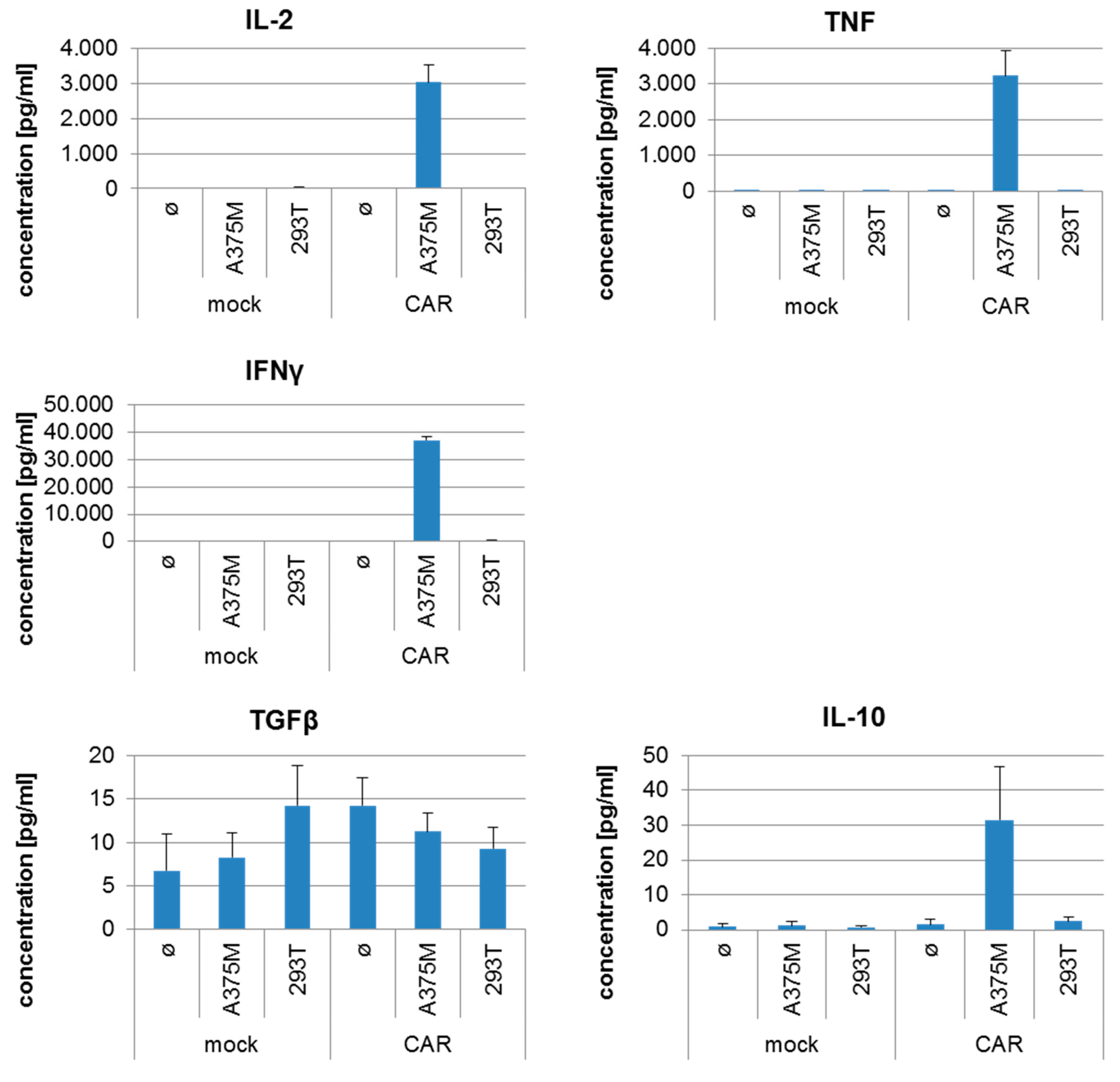
2.4. CSPG4-CAR-Transfected T Cells Show a Very High Potency to Lyse Melanoma Target Cells
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. T-Cell Expansion
4.3. In Vitro Transcription of mRNA
4.4. RNA Electroporation
4.5. Cryoconservation of Electroporated T Cells
4.6. Flow Cytometry
4.7. Cytotoxicity Assay
4.8. Cytokine Secretion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, Y.; Han, W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017, 8, 896–925. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Latchoumanin, O.; Wu, G.; Zhou, G.; Hebbard, L.; George, J.; Qiao, L. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017, 390, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-Cell Therapy for Solid Tumors. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Arabi, F.; Torabi-Rahvar, M.; Shariati, A.; Ahmadbeigi, N.; Naderi, M. Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp. Cell Res. 2018, 369, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Wilson, B.S.; Ruberto, G.; Ferrone, S. Immunochemical characterization of a human high molecular weight--melanoma associated antigen identified with monoclonal antibodies. Cancer Immunol. Immunother. 1983, 14, 196–201. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef]
- Natali, P.G.; Giacomini, P.; Russo, C.; Steinbach, G.; Fenoglio, C.; Ferrone, S. Antigenic profile of human melanoma cells. Analysis with monoclonal antibodies to histocompatibility antigens and to melanoma-associated antigens. J. Cutan. Pathol. 1983, 10, 225–237. [Google Scholar] [CrossRef]
- Li, Y.; Madigan, M.C.; Lai, K.; Conway, R.M.; Billson, F.A.; Crouch, R.; Allen, B.J. Human uveal melanoma expresses NG2 immunoreactivity. Br. J. Ophthalmol. 2003, 87, 629–632. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Rizvi, S.M.; Jager, M.J.; Conway, R.M.; Billson, F.A.; Allen, B.J.; Madigan, M.C. In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4365–4371. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Rooprai, H.K.; Davies, D.; Levine, J.M.; Butt, A.M.; Pilkington, G.J. The NG2 chondroitin sulfate proteoglycan: Role in malignant progression of human brain tumours. Int. J. Dev. Neurosci. 1999, 17, 421–435. [Google Scholar] [CrossRef]
- Godal, A.; Bruland, O.; Haug, E.; Aas, M.; Fodstad, O. Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br. J. Cancer 1986, 53, 839–841. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, Y.; Nishiyama, A.; Chang, A.; Mork, S.; Barnett, G.H.; Cowell, J.K.; Trapp, B.D.; Staugaitis, S.M. Expression of oligodendrocyte progenitor cell antigens by gliomas: Implications for the histogenesis of brain tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 10361–10366. [Google Scholar] [CrossRef] [PubMed]
- Yadavilli, S.; Hwang, E.I.; Packer, R.J.; Nazarian, J. The Role of NG2 Proteoglycan in Glioma. Transl. Oncol. 2016, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Behm, F.G.; Smith, F.O.; Raimondi, S.C.; Pui, C.H.; Bernstein, I.D. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996, 87, 1134–1139. [Google Scholar]
- Hilden, J.M.; Smith, F.O.; Frestedt, J.L.; McGlennen, R.; Howells, W.B.; Sorensen, P.H.; Arthur, D.C.; Woods, W.G.; Buckley, J.; Bernstein, I.D.; et al. MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997, 89, 3801–3805. [Google Scholar]
- Schwartz, S.; Rieder, H.; Schlager, B.; Burmeister, T.; Fischer, L.; Thiel, E. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: Close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003, 17, 1589–1595. [Google Scholar] [CrossRef]
- Smith, F.O.; Rauch, C.; Williams, D.E.; March, C.J.; Arthur, D.; Hilden, J.; Lampkin, B.C.; Buckley, J.D.; Buckley, C.V.; Woods, W.G.; et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996, 87, 1123–1133. [Google Scholar]
- Wuchter, C.; Harbott, J.; Schoch, C.; Schnittger, S.; Borkhardt, A.; Karawajew, L.; Ratei, R.; Ruppert, V.; Haferlach, T.; Creutzig, U.; et al. Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000, 14, 1232–1238. [Google Scholar] [CrossRef]
- Wang, X.; Osada, T.; Wang, Y.; Yu, L.; Sakakura, K.; Katayama, A.; McCarthy, J.B.; Brufsky, A.; Chivukula, M.; Khoury, T.; et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J. Natl. Cancer Inst. 2010, 102, 1496–1512. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.A.; Dallatomasina, A.; Perris, R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015, 5, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, S.; Chen, Z.J.; Liu, C.C.; Hirai, S.; Kageshita, T.; Mittelman, A. Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharm. Ther. 1993, 57, 259–290. [Google Scholar] [CrossRef]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393–1405. [Google Scholar] [PubMed]
- Midwood, K.S.; Salter, D.M. Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthr. Cartil. 1998, 6, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Tordsson, J.M.; Ohlsson, L.G.; Abrahmsen, L.B.; Karlstrom, P.J.; Lando, P.A.; Brodin, T.N. Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol. Immunother. 2000, 48, 691–702. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef]
- Petrini, S.; Tessa, A.; Carrozzo, R.; Verardo, M.; Pierini, R.; Rizza, T.; Bertini, E. Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol. Cell. Neurosci. 2003, 23, 219–231. [Google Scholar] [CrossRef]
- Challier, J.C.; Carbillon, L.; Kacemi, A.; Vervelle, C.; Bintein, T.; Galtier, M.; Espie, M.J.; Uzan, S. Characterization of first trimester human fetal placental vessels using immunocytochemical markers. Cell. Mol. Biol (Noisy-Le-Grand) 2001, 47 Online Pub, OL79–OL87. [Google Scholar]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef]
- Wang, Y.; Geldres, C.; Ferrone, S.; Dotti, G. Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin. Ther. Targets 2015, 19, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- De Vries, J.E.; Keizer, G.D.; te Velde, A.A.; Voordouw, A.; Ruiter, D.; Rumke, P.; Spits, H.; Figdor, C.G. Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int. J. Cancer 1986, 38, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Ozerdem, U. Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006, 66, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Ozerdem, U. Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic. Res. 2006, 38, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, E.; Schmitt, B.M.; Menger, M.D.; Laschke, M.W. The regulatory mechanisms of NG2/CSPG4 expression. Cell. Mol. Biol. Lett. 2017, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Schroff, R.W.; Woodhouse, C.S.; Foon, K.A.; Oldham, R.K.; Farrell, M.M.; Klein, R.A.; Morgan, A.C., Jr. Intratumor localization of monoclonal antibody in patients with melanoma treated with antibody to a 250,000-dalton melanoma-associated antigen. J. Natl. Cancer Inst. 1985, 74, 299–306. [Google Scholar] [PubMed]
- Allen, B.J.; Raja, C.; Rizvi, S.; Li, Y.; Tsui, W.; Graham, P.; Thompson, J.F.; Reisfeld, R.A.; Kearsley, J. Intralesional targeted alpha therapy for metastatic melanoma. Cancer Biol. Ther. 2005, 4, 1318–1324. [Google Scholar] [CrossRef]
- Brehm, H.; Niesen, J.; Mladenov, R.; Stein, C.; Pardo, A.; Fey, G.; Helfrich, W.; Fischer, R.; Gattenlohner, S.; Barth, S. A CSPG4-specific immunotoxin kills rhabdomyosarcoma cells and binds to primary tumor tissues. Cancer Lett. 2014, 352, 228–235. [Google Scholar] [CrossRef]
- Hjortland, G.O.; Garman-Vik, S.S.; Juell, S.; Olsen, O.E.; Hirschberg, H.; Fodstad, O.; Engebraaten, O. Immunotoxin treatment targeted to the high-molecular-weight melanoma-associated antigen prolonging the survival of immunodeficient rats with invasive intracranial human glioblastoma multiforme. J. Neurosurg. 2004, 100, 320–327. [Google Scholar] [CrossRef]
- Schwenkert, M.; Birkholz, K.; Schwemmlein, M.; Kellner, C.; Kugler, M.; Peipp, M.; Nettelbeck, D.M.; Schuler-Thurner, B.; Schaft, N.; Dorrie, J.; et al. A single chain immunotoxin, targeting the melanoma-associated chondroitin sulfate proteoglycan, is a potent inducer of apoptosis in cultured human melanoma cells. Melanoma Res. 2008, 18, 73–84. [Google Scholar] [CrossRef]
- Jordaan, S.; Chetty, S.; Mungra, N.; Koopmans, I.; van Bommel, P.E.; Helfrich, W.; Barth, S. CSPG4: A Target for Selective Delivery of Human Cytolytic Fusion Proteins and TRAIL. Biomedicines 2017, 5, 37. [Google Scholar] [CrossRef]
- De Bruyn, M.; Rybczynska, A.A.; Wei, Y.; Schwenkert, M.; Fey, G.H.; Dierckx, R.A.; van Waarde, A.; Helfrich, W.; Bremer, E. Melanoma-associated Chondroitin Sulfate Proteoglycan (MCSP)-targeted delivery of soluble TRAIL potently inhibits melanoma outgrowth in vitro and in vivo. Mol. Cancer 2010, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef]
- Geldres, C.; Savoldo, B.; Hoyos, V.; Caruana, I.; Zhang, M.; Yvon, E.; Del, V.M.; Creighton, C.J.; Ittmann, M.; Ferrone, S.; et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res. 2014, 20, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Abken, H.; Hombach, A.; Heuser, C.; Reinhold, U. A novel strategy in the elimination of disseminated melanoma cells: Chimeric receptors endow T cells with tumor specificity. Recent Results Cancer Res. 2001, 158, 249–264. [Google Scholar] [PubMed]
- Burns, W.R.; Zhao, Y.; Frankel, T.L.; Hinrichs, C.S.; Zheng, Z.; Xu, H.; Feldman, S.A.; Ferrone, S.; Rosenberg, S.A.; Morgan, R.A. A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res. 2010, 70, 3027–3033. [Google Scholar] [CrossRef]
- Losch, F.O.; Muller, R.; Mutschler, B.; Neri, D.; Natali, P.G.; Reth, M.; Carsetti, R. Activation of T cells via tumor antigen specific chimeric receptors: The role of the intracellular signaling domain. Int. J. Cancer 2003, 103, 399–407. [Google Scholar] [CrossRef]
- Reinhold, U.; Liu, L.; Ludtke-Handjery, H.C.; Heuser, C.; Hombach, A.; Wang, X.; Tilgen, W.; Ferrone, S.; Abken, H. Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J. Investig. Derm. 1999, 112, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.; Kopecky, C.; Hombach, A.; Zigrino, P.; Mauch, C.; Abken, H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 2474–2479. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Savoldo, B.; Di, I.N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, eaao2731. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef]
- Dorrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Schaft, N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol. Immunother. 2014, 63, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.; Eirikis, E.; Davis, C.; Davis, H.M.; Prabhakar, U. Comparative analysis of lymphocyte activation marker expression and cytokine secretion profile in stimulated human peripheral blood mononuclear cell cultures: An in vitro model to monitor cellular immune function. J. Immunol. Methods 2004, 293, 127–142. [Google Scholar] [CrossRef]
- Barrett, D.M.; Singh, N.; Porter, D.L.; Grupp, S.A.; June, C.H. Chimeric antigen receptor therapy for cancer. Annu. Rev. Med. 2014, 65, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; June, C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef]
- Dai, X.; Mei, Y.; Cai, D.; Han, W. Standardizing CAR-T therapy: Getting it scaled up. Biotechnol. Adv. 2019, 37, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Riviere, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, Z.; Robbins, P.F.; Khong, H.T.; Rosenberg, S.A.; Morgan, R.A. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J. Immunol. 2005, 174, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.; Khong, H.T.; Rosenberg, S.A.; Morgan, R.A. Transduction of an HLA-DP4-restricted NY-ESO-1-specific TCR into primary human CD4+ lymphocytes. J. Immunother. 2006, 29, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Birkholz, K.; Hombach, A.; Krug, C.; Reuter, S.; Kershaw, M.; Kampgen, E.; Schuler, G.; Abken, H.; Schaft, N.; Dorrie, J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009, 16, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific Chimeric Antigen Receptor mRNA-Engineered T cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Kulikovskaya, I.; Suhoski, M.M.; Melenhorst, J.J.; Loudon, B.; Mato, A.R.; et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood 2018, 132, 1022–1026. [Google Scholar] [CrossRef]
- Noto, A.; Ngauv, P.; Trautmann, L. Cell-based flow cytometry assay to measure cytotoxic activity. J. Vis. Exp. 2013, e51105. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | 2 PBMCs Day 0 | Cell Number Harvested at Day 9 | Maximum Number of Cells Electro-Porated at Day 9 | CAR-Transfected T Cells at Day 9 | Survival after Electro-Poration | Survival after Electroporation and Cryo-Conservation | Yield Factor |
|---|---|---|---|---|---|---|---|
| Con 1 | 292 | 4876 | 3240 | 2278 | 70.3% | 55.1% | 7.8 |
| Con 2 | 231 | 3996 | 3240 | 2456 | 75.8% | 61.4% | 10.6 |
| Con 3 | 85 | 5466 | 3240 | 2450 | 75.6% | 61.4% | 28.8 |
| Con 4 | 341 | 4344 | 3240 | 2424 | 74.8% | 55.6% | 7.1 |
| Average | 237.3 | 4670.5 | 3240 | 2402 | 74.1% | 58.4% | 13.6 |
| SD | 111.0 | 642.0 | 83.8 | 2.6% | 3.5% | 10.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiesinger, M.; März, J.; Kummer, M.; Schuler, G.; Dörrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers 2019, 11, 1198. https://doi.org/10.3390/cancers11081198
Wiesinger M, März J, Kummer M, Schuler G, Dörrie J, Schuler-Thurner B, Schaft N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers. 2019; 11(8):1198. https://doi.org/10.3390/cancers11081198
Chicago/Turabian StyleWiesinger, Manuel, Johannes März, Mirko Kummer, Gerold Schuler, Jan Dörrie, Beatrice Schuler-Thurner, and Niels Schaft. 2019. "Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance" Cancers 11, no. 8: 1198. https://doi.org/10.3390/cancers11081198
APA StyleWiesinger, M., März, J., Kummer, M., Schuler, G., Dörrie, J., Schuler-Thurner, B., & Schaft, N. (2019). Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers, 11(8), 1198. https://doi.org/10.3390/cancers11081198