HMGA1 Modulates Gene Transcription Sustaining a Tumor Signalling Pathway Acting on the Epigenetic Status of Triple-Negative Breast Cancer Cells


Abstract
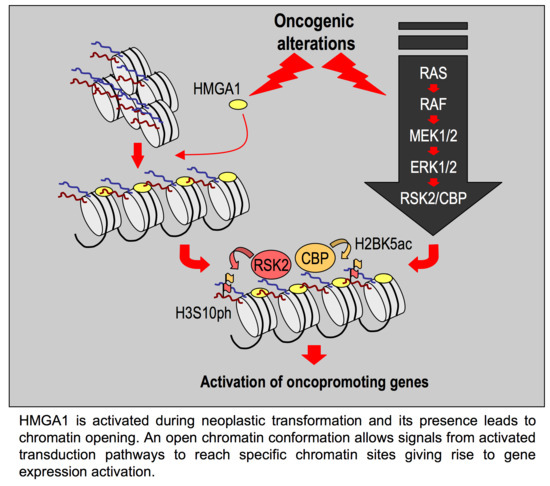
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
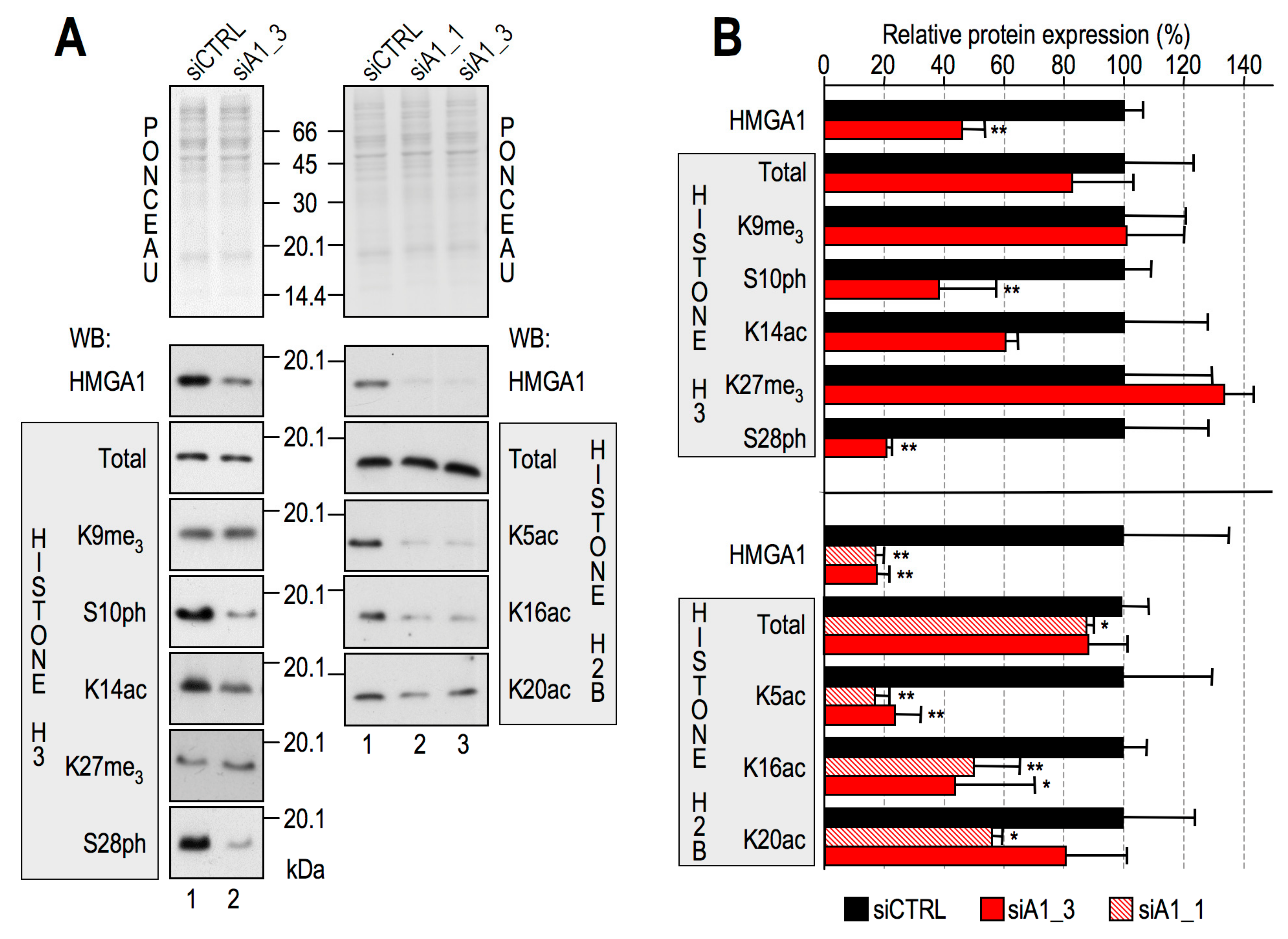
2.1. HMGA1 Expression Influences the Histone Code
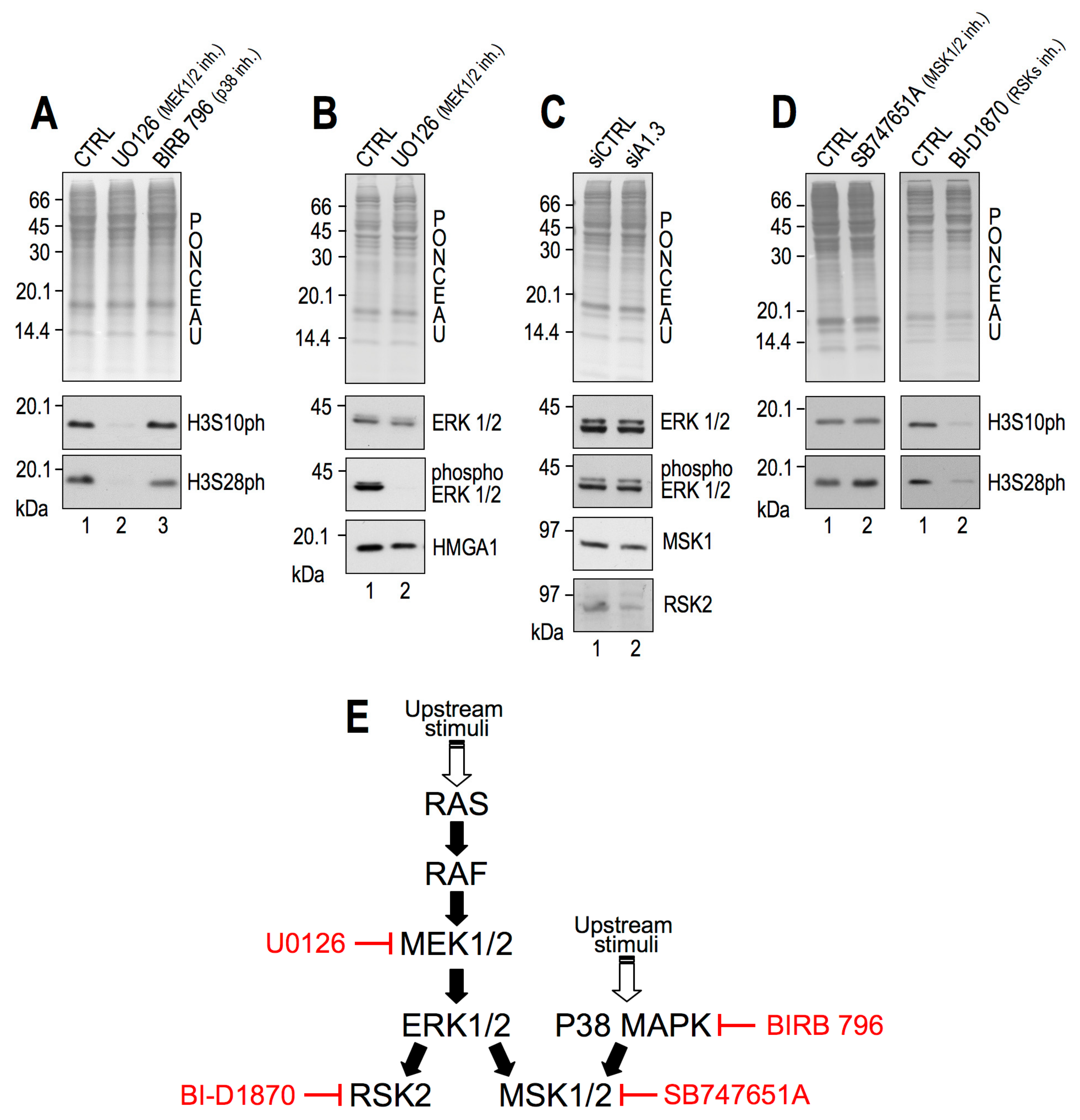
2.2. RSK2 Kinase Phosphorylates H3S10 and H3S28 in Interphasic MDA-MB–231 Cells
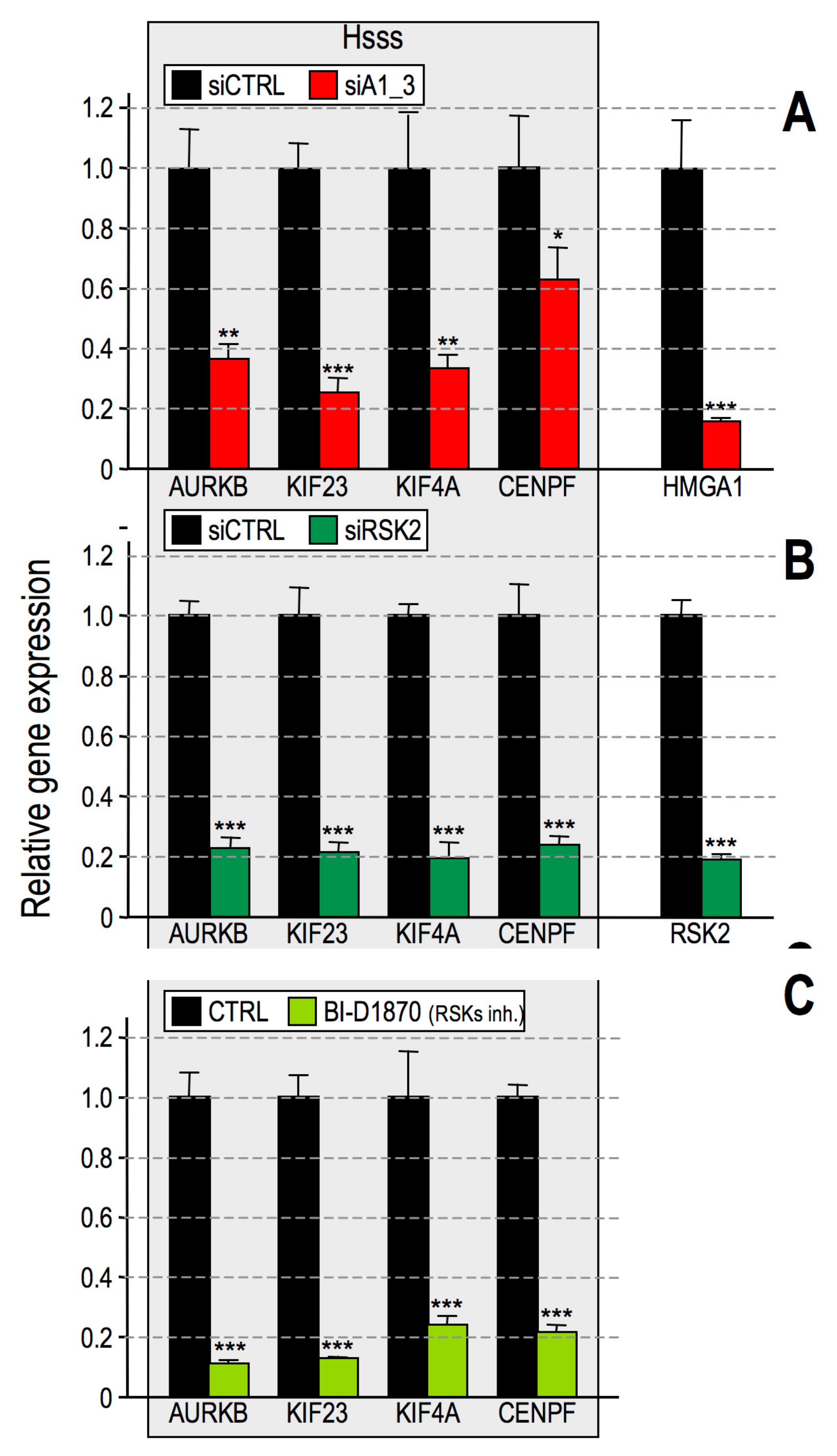
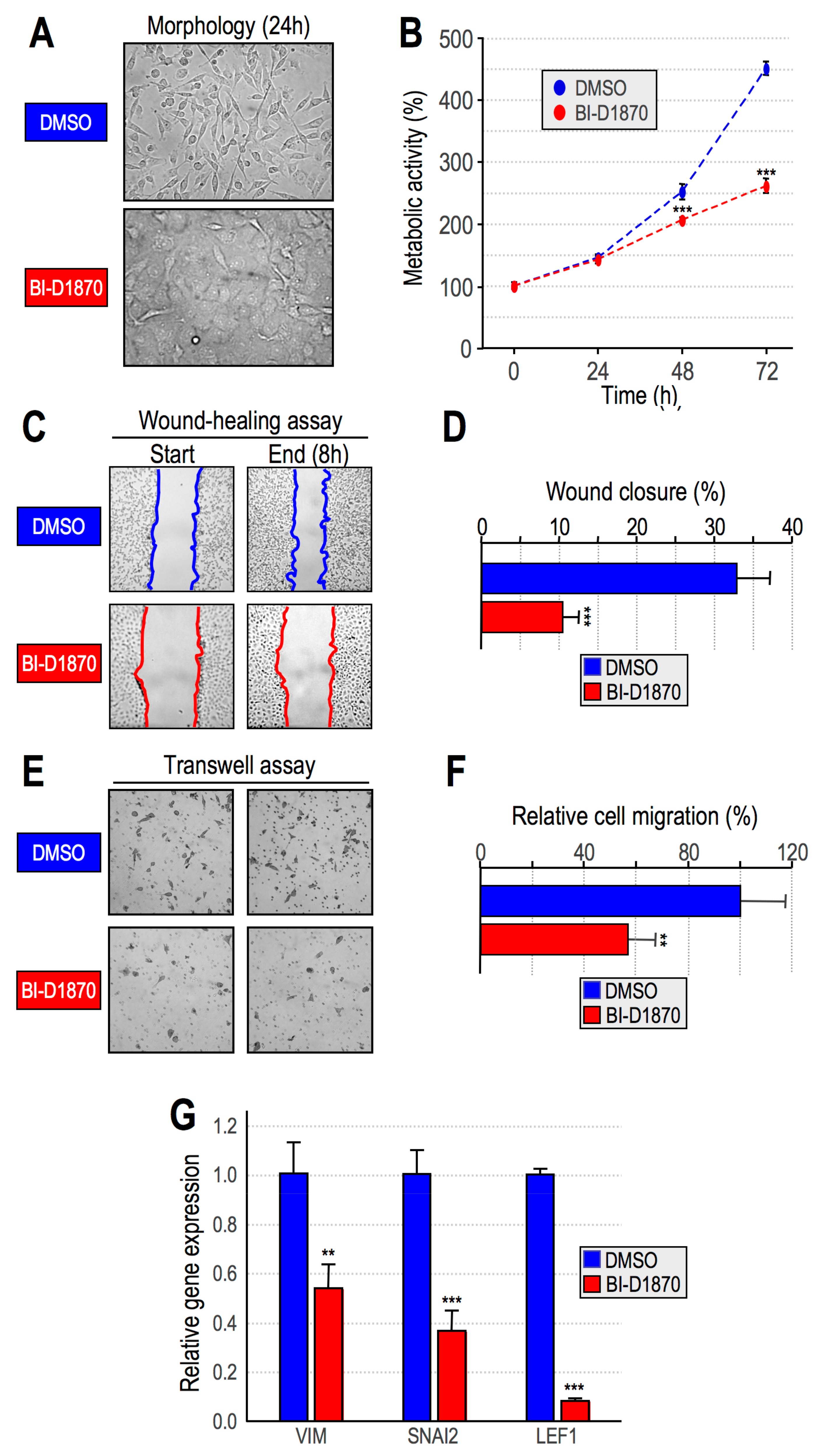
2.3. RSK2 Impairment Mimics the Effects of HMGA1 Silencing
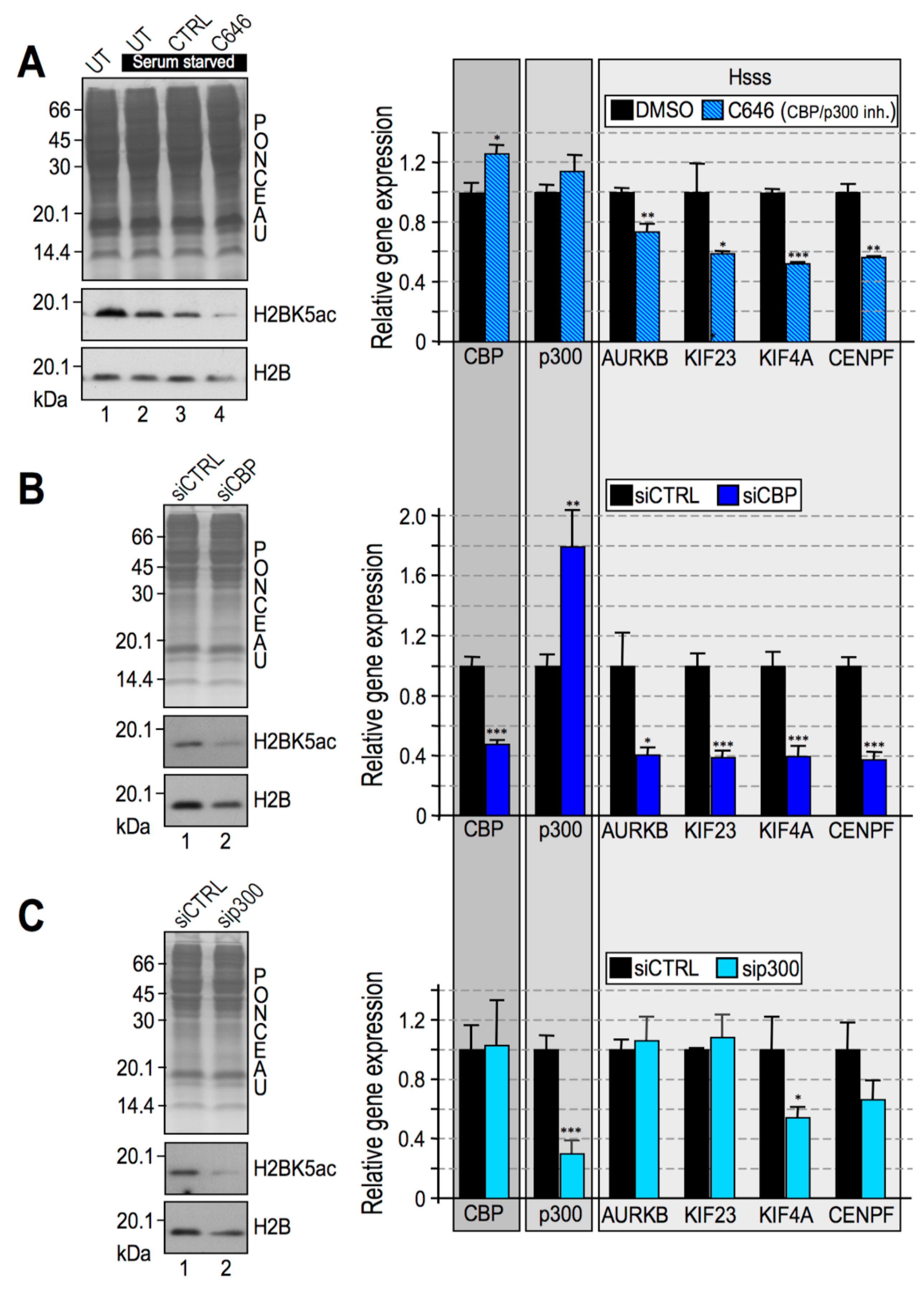
2.4. CBP Impairment Mimics the Effects of HMGA1 Silencing
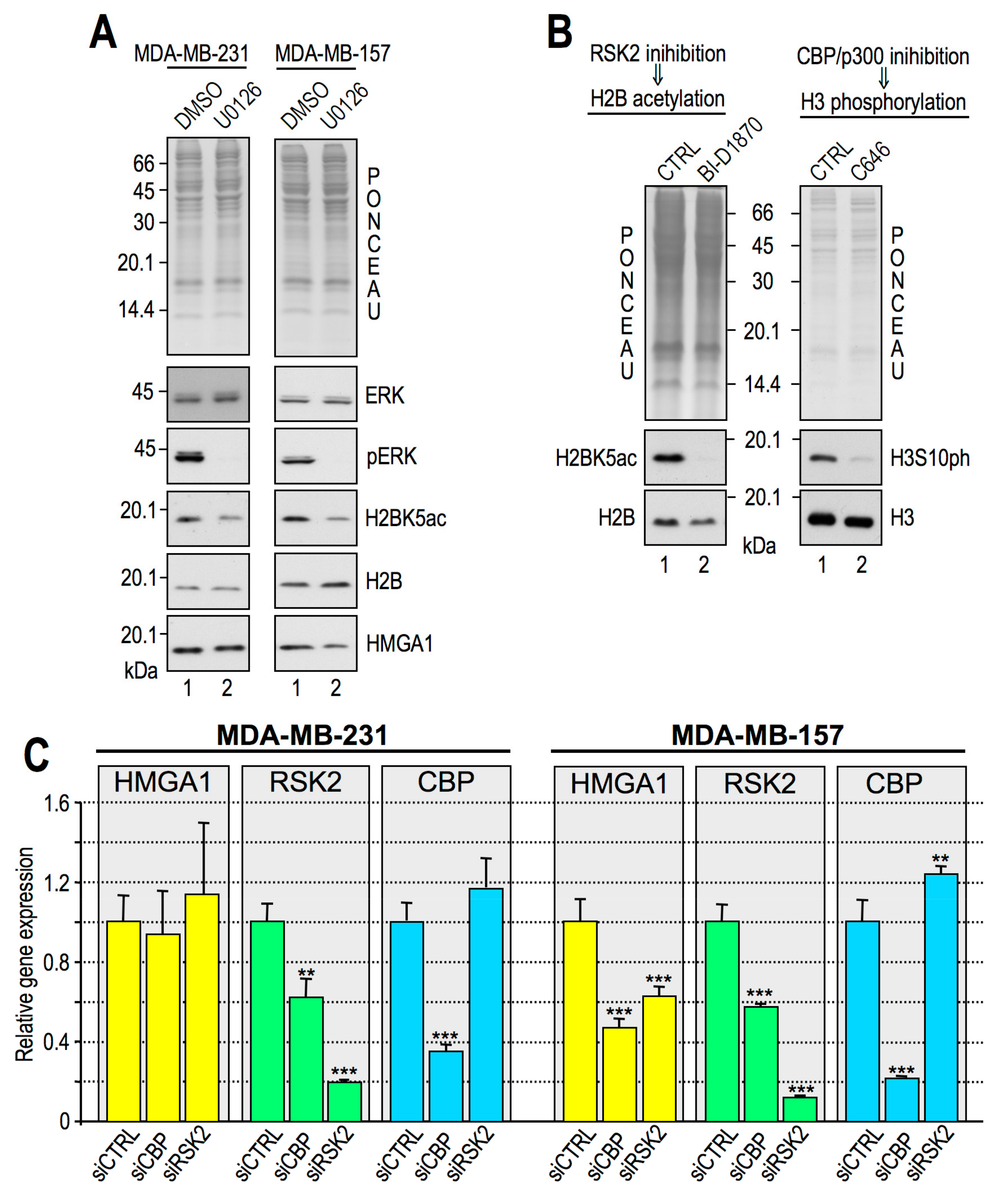
2.5. Histone H3S10ph and Histone H2BK5ac are Interdependent PTMs
2.6. HMGA1 Modulation of RSK2 and CBP Activities Is Likely to Occur by a Direct Mechanism and Not by Affecting Their Gene or Protein Expression Levels
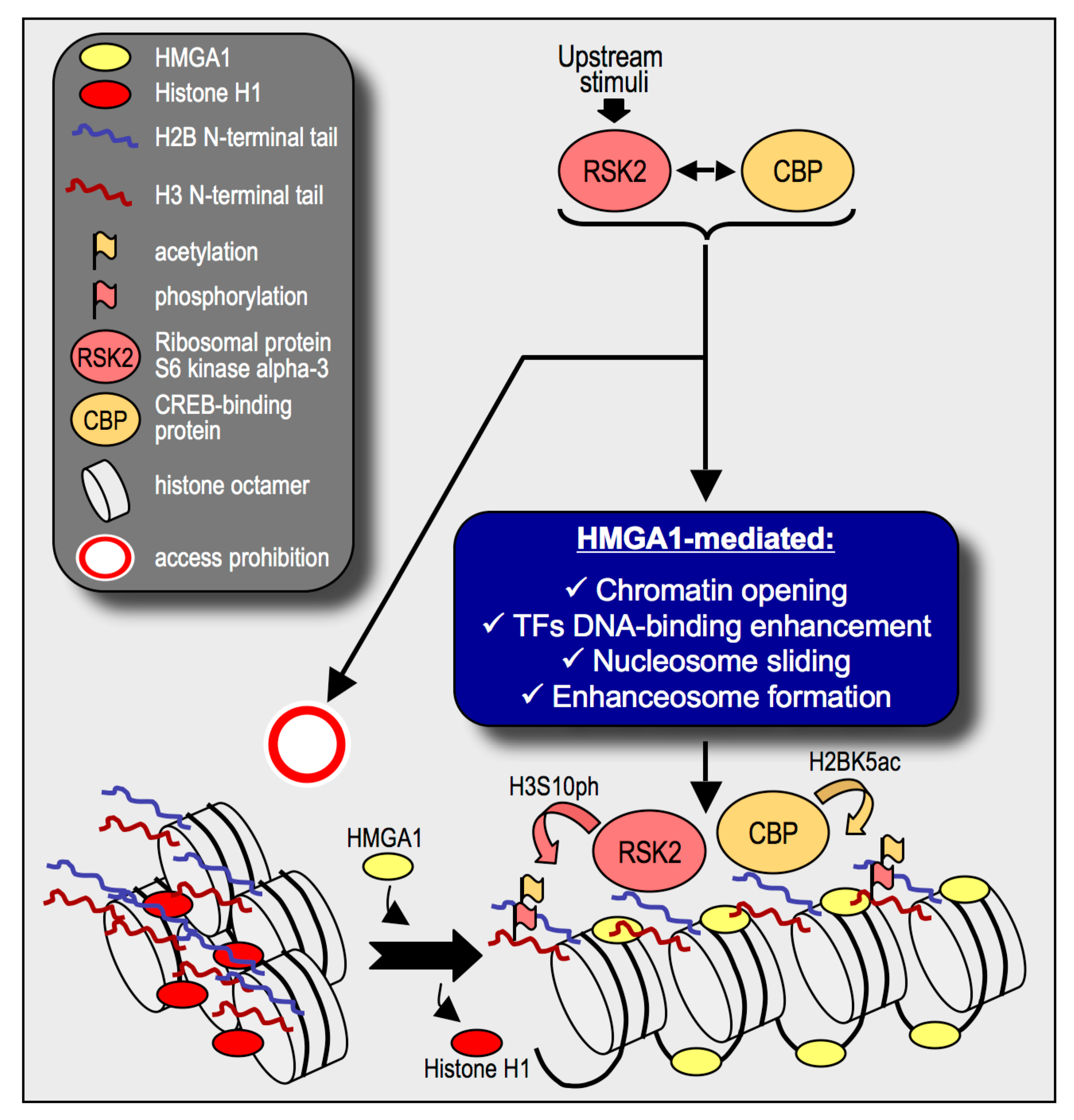
3. Discussion
- (a)
- HMGA1 silencing causes the downregulation of a series of histone post-translational modifications, i.e., mainly histone H3 S10 and S28 phosphorylation and histone H2B K5, K16, and K20 acetylation;
- (b)
- In the MDA-MB-231 cellular model these modification are due to the activity of RSK2 and CBP/p300;
- (c)
- The silencing of RSK2 and CBP phenocopies the effect of HMGA1 silencing towards a set of HMGA1 regulated genes while p300 silencing does not;
- (d)
- Impairment of RSK2 activity by means of the RSKs inhibitor BI-D1870 is responsible for a decrease of histone H3 S10 phosphorylation at specific regulatory regions of HMGA1 regulated genes;
- (e)
- HMGA1 does not modulate the gene/protein expression level of RSK2 and CBP and HMGA1 does not modulate the nuclear localization of RSK2 neither its activation status (i.e., RSK2 S227 phosphorylation level);
- (f)
- Impairment of RSK2 activity by means of the RSKs inhibitor BI-D1870 is responsible for MET;
- (g)
- HMGA1 and RSK2 interact in a DNA-dependent way.
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. qRT-PCR, Cell proliferation (MTS) assay, Wound Healing Assay, Transwell Migration Assay, SDS-PAGE, and Western Blot Analyses
4.3. Immunofluorescence (IF) Analyses
4.4. Co-immunoprecipitation Assay
4.5. Chromatin Immunoprecipitation
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Nunez, F.J.; Mendez, F.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lowenstein, P.R.; Castro, M.G. Mutated chromatin regulatory factors as tumor drivers in cancer. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, M.; Holth, L.T.; Zoghbi, H.Y.; Reeves, R. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Lehn, D.A.; Reeves, R. Alternative processing of mRNAs encoding mammalian chromosomal high-mobility-group proteins HMG-I and HMG-Y. Mol. Cell. Biol. 1989, 9, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Chau, K.Y.; Patel, U.A.; Lee, K.L.; Lam, H.Y.; Crane-Robinson, C. The gene for the human architectural transcription factor HMGI-C consists of five exons each coding for a distinct functional element. Nucleic Acids Res. 1995, 23, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, G.; Avantaggiato, V.; Visconti, R.; Fedele, M.; Battista, S.; Trapasso, F.; Merciai, B.M.; Fidanza, V.; Giancotti, V.; Santoro, M.; et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene 1996, 13, 2439–2446. [Google Scholar] [PubMed]
- Lund, T.; Holtlund, J.; Fredriksen, M.; Laland, S.G. On the presence of two new high mobility group-like proteins in HeLa S3 cells. FEBS Lett. 1983. [Google Scholar] [CrossRef]
- Giancotti, V.; Berlingieri, M.T.; Difiore, P.P.; Fusco, A.; Vecchio, G.; Crane-Robinson, C. Changes in Nuclear Proteins on Transformation of Rat Epithelial Thyroid Cells by a Murine Sarcoma Retrovirus. Cancer Res. 1985, 45, 6051–6057. [Google Scholar]
- Giancotti, V.; Buratti, E.; Perissin, L.; Zorzet, S.; Balmain, A.; Portella, G.; Fusco, A.; Goodwin, G.H. Analysis of the HMGI nuclear proteins in mouse neoplastic cells induced by different procedures. Exp. Cell Res. 1989. [Google Scholar] [CrossRef]
- Vallone, D.; Battista, S.; Pierantoni, G.M.; Fedele, M.; Casalino, L.; Santoro, M.; Viglietto, G.; Fusco, A.; Verde, P. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J. 1997. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.J.; Mukherjee, M.; Dolde, C.E.; Xu, Y.; Maher, J.F.; Bunton, T.E.; Williams, J.B.; Resar, L.M. HMG-I/Y, a new c-Myc target gene and potential oncogene. Mol. Cell. Biol. 2000, 20, 5490–5502. [Google Scholar] [CrossRef] [PubMed]
- Battista, S.; Fidanza, V.; Fedele, M.; Klein-Szanto, A.J.P.; Outwater, E.; Brunner, H.; Santoro, M.; Croce, C.M.; Fusco, A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999, 59, 4793–4797. [Google Scholar] [PubMed]
- Fedele, M.; Pentimalli, F.; Baldassarre, G.; Battista, S.; Klein-Szanto, A.J.P.; Kenyon, L.; Visone, R.; De Martino, I.; Ciarmiello, A.; Arra, C.; et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005, 24, 3427–3435. [Google Scholar] [CrossRef] [PubMed]
- Yie, J.; Merika, M.; Munshi, N.; Chen, G.; Thanos, D. The role of HMG I(Y) in the assembly and function of the IFN-β enhanceosome. EMBO J. 1999. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Agalioti, T.; Lomvardas, S.; Merika, M.; Chen, G.; Thanos, D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science 2001. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Rustighi, A.; Malaguarnera, R.; Altamura, S.; Vigneri, P.; Del Sal, G.; Giancotti, V.; Pezzino, V.; Vigneri, R.; Manfioletti, G. HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res. 2006, 66, 2980–2989. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006. [Google Scholar] [CrossRef]
- Ueda, Y.; Watanabe, S.; Tei, S.; Saitoh, N.; Kuratsu, J.I.; Nakao, M. High mobility group protein HMGA1 inhibits retinoblastoma protein-mediated cellular G0 arrest. Cancer Sci. 2007, 98, 1893–1901. [Google Scholar] [CrossRef]
- Sgarra, R.; Furlan, C.; Zammitti, S.; Lo Sardo, A.; Maurizio, E.; Di Bernardo, J.; Giancotti, V.; Manfioletti, G. Interaction proteomics of the HMGA chromatin architectural factors. Proteomics 2008. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Tessari, M.A.; Di Bernardo, J.; Rustighi, A.; Zago, P.; Liberatori, S.; Armini, A.; Bini, L.; Giancotti, V.; Manfioletti, G. Discovering high mobility group A molecular partners in tumour cells. Proteomics 2005. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Zammitti, S.; Lo Sardo, A.; Maurizio, E.; Arnoldo, L.; Pegoraro, S.; Giancotti, V.; Manfioletti, G. HMGA molecular network: From transcriptional regulation to chromatin remodeling. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Nissen, M.S. Interaction of high mobility group-I (Y) nonhistone proteins with nucleosome core particles. J. Biol. Chem. 1993, 268, 21137–21146. [Google Scholar] [PubMed]
- Sgarra, R.; Pegoraro, S.; Ros, G.; Penzo, C.; Chiefari, E.; Foti, D.; Brunetti, A.; Manfioletti, G. High Mobility Group A (HMGA) proteins: Molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, C.; Li, T.; Ding, Y.; Tu, T.; Zhou, F.; Qi, W.; Chen, H.; Sun, X. Let-7a inhibits growth and migration of breast cancer cells by targeting HMGA1. Int. J. Oncol 2015, 46, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Bin Zhou, W.; Zhong, C.N.; Luo, X.P.; Zhang, Y.Y.; Zhang, G.Y.; Zhou, D.X.; Liu, L.P. MIR-625 suppresses cell proliferation and migration by targeting HMGA1 in breast cancer. Biochem. Biophys. Res. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Maurizio, E.; Wiśniewski, J.R.; Ciani, Y.; Amato, A.; Arnoldo, L.; Penzo, C.; Pegoraro, S.; Giancotti, V.; Zambelli, A.; Piazza, S.; et al. Translating Proteomic Into Functional Data: An High Mobility Group A1 (HMGA1) Proteomic Signature Has Prognostic Value in Breast Cancer. Mol. Cell. Proteomics 2016, 15, 109–123. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef]
- Reeves, R.; Edberg, D.D.; Li, Y. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell. Biol. 2001, 21, 575–594. [Google Scholar] [CrossRef]
- Treff, N.R.; Pouchnik, D.; Dement, G.A.; Britt, R.L.; Reeves, R. High-mobility group A1a protein regulates Ras/ERK signaling in MCF-7 human breast cancer cells. Oncogene 2004, 23, 777–785. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pellarin, I.; Arnoldo, L.; Costantini, S.; Pegoraro, S.; Ros, G.; Penzo, C.; Triolo, G.; Demarchi, F.; Sgarra, R.; Vindigni, A.; et al. The architectural chromatin factor high mobility group A1 enhancesdna Ligase IV activity influencingdna repair. PLoS ONE 2016, 11, e0164258. [Google Scholar] [CrossRef] [PubMed]
- Ohe, K.; Miyajima, S.; Abe, I.; Tanaka, T.; Hamaguchi, Y.; Harada, Y.; Horita, Y.; Beppu, Y.; Ito, F.; Yamasaki, T.; et al. HMGA1a induces alternative splicing of estrogen receptor alpha in MCF-7 human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Resmini, G.; Rizzo, S.; Franchin, C.; Zanin, R.; Penzo, C.; Pegoraro, S.; Ciani, Y.; Piazza, S.; Arrigoni, G.; Sgarra, R.; et al. HMGA1 regulates the Plasminogen activation system in the secretome of breast cancer cells. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Senigagliesi, B.; Penzo, C.; Severino, L.U.; Maraspini, R.; Petrosino, S.; Morales-Navarrete, H.; Pobega, E.; Ambrosetti, E.; Parisse, P.; Pegoraro, S.; et al. The High Mobility Group A1 (HMGA1) chromatin architectural factor modulates nuclear stiffness in breast cancer cells. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Mendez, O.; Peg, V.; Salvans, C.; Pujals, M.; Fernandez, Y.; Abasolo, I.; Perez, J.; Matres, A.; Valeri, M.; Gregori, J.; et al. Extracellular HMGA1 promotes tumor invasion and metastasis in triple-negative breast cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Sgarra, R.; Rustighi, A.; Tessari, M.A.; Di Bernardo, J.; Altamura, S.; Fusco, A.; Manfioletti, G.; Giancotti, V. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 2004, 574, 1–8. [Google Scholar] [CrossRef]
- Sabbattini, P.; Sjoberg, M.; Nikic, S.; Frangini, A.; Holmqvist, P.-H.; Kunowska, N.; Carroll, T.; Brookes, E.; Arthur, S.J.; Pombo, A.; et al. An H3K9/S10 methyl-phospho switch modulates Polycomb and Pol II binding at repressed genes during differentiation. Mol. Biol. Cell 2014. [Google Scholar] [CrossRef]
- Winter, S.; Simboeck, E.; Fischle, W.; Zupkovitz, G.; Dohnal, I.; Mechtler, K.; Ammerer, G.; Seiser, C. 14-3-3 Proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J. 2008. [Google Scholar] [CrossRef]
- Lau, P.N.I.; Cheung, P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef]
- Agalioti, T.; Chen, G.; Thanos, D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002. [Google Scholar] [CrossRef]
- Parra, M.A.; Kerr, D.; Fahy, D.; Pouchnik, D.J.; Wyrick, J.J. Deciphering the Roles of the Histone H2B N-Terminal Domain in Genome-Wide Transcription. Mol. Cell. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, H.; Kim, K.; Lee, B.; Lu, W.; An, W. Selective requirement of H2B N-Terminal tail for p14ARF-induced chromatin silencing. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes Dev. 2002. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Boyle, S.; Sutherland, H.; De Las Heras, J.; Allan, J.; Jenuwein, T.; Bickmore, W.A. Formation of facultative heterochromatin in the absence of HP1. EMBO J. 2003. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.F.M.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.H.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin. Mol. Cell 2003. [Google Scholar] [CrossRef]
- Thomson, S.; Clayton, A.L.; Hazzalin, C.A.; Rose, S.; Barratt, M.J.; Mahadevan, L.C. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999. [Google Scholar] [CrossRef]
- Zhong, S.P.; Ma, W.Y.; Dong, Z. ERKs and p38 kinases mediate ultraviolet B-induced phosphorylation of histone H3 at serine 10. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Sassone-Corsi, P.; Mizzen, C.A.; Cheung, P.; Crosio, C.; Monaco, L.; Jacquot, S.; Hanauer, A.; Allis, C.D. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science 1999, 285, 886–891. [Google Scholar] [CrossRef]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Márquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005. [Google Scholar] [CrossRef]
- Favata, M.F. Identification of a Novel Inhibitor of Mitogen-activated Protein Kinase Kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.; Macdonald, A.; McCoy, C.E.; Darragh, J.; Reith, A.D.; Arthur, J.S.C. Characterization of the cellular action of the MSK inhibitor SB-747651A. Biochem. J. 2011. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, G.P.; Cummings, L.; Newell, F.S.; Armstrong, C.; Bain, J.; Frodin, M.; Grauert, M.; Hoffmann, M.; Schnapp, G.; Steegmaier, M.; et al. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 2006. [Google Scholar] [CrossRef]
- Pegoraro, S.; Ros, G.; Ciani, Y.; Sgarra, R.; Piazza, S.; Manfioletti, G. A novel HMGA1-CCNE2-YAP axis regulates breast cancer aggressiveness. Oncotarget 2015, 6, 19087–19101. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [PubMed]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.A.; Kuo, Y.M.; Bhattacharjee, V.; Yen, T.J.; Andrews, A.J. Changing the selectivity of p300 by acetyl-coa modulation of histone acetylation. ACS Chem. Biol. 2015, 10, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Merienne, K.; Pannetier, S.; Harel-Bellan, A.; Sassone-Corsi, P. Mitogen-Regulated RSK2-CBP Interaction Controls Their Kinase and Acetylase Activities. Mol. Cell. Biol. 2001, 21, 7089–7096. [Google Scholar] [CrossRef]
- Jensen, C.J.; Buch, M.B.; Krag, T.O.; Hemmings, B.A.; Gammeltoft, S.; Frödin, M. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3- phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 1999, 274, 27168–27176. [Google Scholar] [CrossRef]
- Chen, X.; Hu, H.; He, L.; Yu, X.; Liu, X.; Zhong, R.; Shu, M. A novel subtype classification and risk of breast cancer by histone modification profiling. Breast Cancer Res. Treat. 2016, 157, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Huang, D.; Dai, W.; Yang, F. Overexpression of HMGA1 correlates with the malignant status and prognosis of breast cancer. Mol. Cell. Biochem. 2015, 404, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.; Wolffe, A.P. Substrate Structure Influences Binding of the Non-Histone Protein HMG-I(Y) to Free and Nucleosomal DNA. Biochemistry 2002. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Sahai, V.; Ebine, K.; Kumar, K.; Munshi, H.G. Three-Dimensional Collagen I Promotes Gemcitabine Resistance In Vitro in Pancreatic Cancer Cells through HMGA2-Dependent Histone Acetyltransferase Expression. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.J.; Kahata, K.; Idås, O.; Thuault, S.; Heldin, C.H.; Moustakas, A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015, 43, 162–178. [Google Scholar] [CrossRef]
- Fasci, D.; van Ingen, H.; Scheltema, R.A.; Heck, A.J.R. Histone Interaction Landscapes Visualized by Crosslinking Mass Spectrometry in Intact Cell Nuclei. Mol. Cell. Proteomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, A.; Seiser, C. Histone H3 phosphorylation—A versatile chromatin modification for different occasions. Biochimie 2012, 94, 2193–2201. [Google Scholar] [CrossRef]
- Ludwik, K.; Campbell, J.; Li, M.; Li, Y.; Sandusky, Z.; Pasic, L.; Sowder, M.; Brenin, D.; Pietenpol, J.; O’Doherty, G.; et al. Development of a RSK Inhibitor as a Novel Therapy for Triple Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 2598–2608. [Google Scholar] [CrossRef]
- Stratford, A.L.; Reipas, K.; Hu, K.; Fotovati, A.; Brough, R.; Frankum, J.; Takhar, M.; Watson, P.; Ashworth, A.; Lord, C.J.; et al. Targeting p90 ribosomal S6 kinase eliminates tumor-initiating cells by inactivating Y-box binding protein-1 in triple-negative breast cancers. Stem Cells 2012, 30, 1338–1348. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Marshall, C.B.; Li, G.Y.; Yamamoto, K.; Mak, T.W.; Ikura, M. Synergistic interplay between promoter recognition and CBP/p300 coactivator recruitment by FOXO3a. ACS Chem. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2002. [Google Scholar] [CrossRef] [PubMed]
- Vervoorts, J.; Lüscher-Firzlaff, J.M.; Rottmann, S.; Lilischkis, R.; Walsemann, G.; Dohmann, K.; Austen, M.; Lüscher, B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Sun, J.; Dowhan, D.H.; Ishii, S.; Gonda, T.J. Mutations in Multiple Domains of c-Myb Disrupt Interaction with CBP/p300 and Abrogate Myeloid Transforming Ability. Mol. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; Di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol. Cell. Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Yang, H.; Pinello, C.E.; Luo, J.; Li, D.; Wang, Y.; Zhao, L.Y.; Jahn, S.C.; Saldanha, S.A.; Chase, P.; Planck, J.; et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Mol. Cancer Ther. 2013, 12, 610–620. [Google Scholar] [CrossRef]
- Lawrence, M.C.; McGlynn, K.; Shao, C.; Duan, L.; Naziruddin, B.; Levy, M.F.; Cobb, M.H. Chromatin-bound mitogen-activated protein kinases transmit dynamic signals in transcription complexes in beta-cells. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell. Mol. Life Sci. 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- Thanos, D.; Maniatis, T. The High Mobility Group protein HMG I(Y) is required for NF-κB-dependent virus induction of the human IFN-β gene. Cell 1992. [Google Scholar] [CrossRef]
- Chin, M.T.; Pellacani, A.; Wang, H.; Lin, S.S.J.; Jain, M.K.; Perrella, M.A.; Lee, M.E. Enhancement of serum-response factor-dependent transcription and DNA binding by the architectural transcription factor HMG-I(Y). J. Biol. Chem. 1998. [Google Scholar] [CrossRef] [PubMed]
- Bagga, R.; Michalowski, S.; Sabnis, R.; Griffith, J.D.; Emerson, B.M. HMG I/Y regulates long-range enhancer-dependent transcription on DNA and chromatin by changes in DNA topology. Nucleic Acids Res. 2000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Y. HMG modifications and nuclear function. Biochim. Biophys. Acta Gene Regul. Mech. 2010. [Google Scholar] [CrossRef] [PubMed]
- Sgarra, R.; Diana, F.; Rustighi, A.; Manfioletti, G.; Giancotti, V. Increase of HMGA1a protein methylation is a distinctive characteristic of leukaemic cells induced to undergo apoptosis. Cell Death Differ. 2003. [Google Scholar] [CrossRef]
- Diana, F.; Sgarra, R.; Manfioletti, G.; Rustighi, A.; Poletto, D.; Sciortino, M.T.; Mastino, A.; Giancotti, V. A Link between Apoptosis and Degree of Phosphorylation of High Mobility Group A1a Protein in Leukemic Cells. J. Biol. Chem. 2001. [Google Scholar] [CrossRef]
- Harrer, M.; Lührs, H.; Bustin, M.; Scheer, U.; Hock, R. Dynamic interaction of HMGA1a proteins with chromatin. J. Cell Sci. 2004, 117, 3459–3471. [Google Scholar] [CrossRef]
- Giancotti, V.; Bandiera, A.; Ciani, L.; Santoro, D.; Crane-Robinson, C.; Goodwin, G.H.; Boiocchi, M.; Dolcetti, R.; Casetta, B. High mobility group (HMG) proteins and histone H1 subtypes expression in normal and tumor tissues of mouse. Eur. J. Biochem. 1993. [Google Scholar] [CrossRef]
- Ndlovu, M.N.; Van Lint, C.; Van Wesemael, K.; Callebert, P.; Chalbos, D.; Haegeman, G.; Vanden Berghe, W. Hyperactivated NF- B and AP-1 Transcription Factors Promote Highly Accessible Chromatin and Constitutive Transcription across the Interleukin-6 Gene Promoter in Metastatic Breast Cancer Cells. Mol. Cell. Biol. 2009. [Google Scholar] [CrossRef]
- Vicent, G.P.; Ballaré, C.; Nacht, A.S.; Clausell, J.; Subtil-Rodríguez, A.; Quiles, I.; Jordan, A.; Beato, M. Induction of Progesterone Target Genes Requires Activation of Erk and Msk Kinases and Phosphorylation of Histone H3. Mol. Cell 2006. [Google Scholar] [CrossRef]
- Teng, H.; Ballim, R.D.; Mowla, S.; Prince, S. Phosphorylation of histone H3 by protein kinase C signaling plays a critical role in the regulation of the developmentally important TBX2 gene. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Yumi, Y.; Verma, U.N.; Prajapati, S.; Youn-Tae, K.; Gaynor, R.B. Histone H3 phosphorylation by ikk-alpha is critical for cytokine-induced gene expression. Nature 2003. [Google Scholar] [CrossRef]
- Anest, V.; Hanson, J.L.; Cogswell, P.C.; Steinbrecher, K.A.; Strahl, B.D.; Baldwin, A.S. A nucleosomal function for ikappab kinase-alpha in nf-kappaB-dependent gene expression. Nature 2003. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hung, M.C. Beyond NF-κB activation: Nuclear functions of IκB kinase α. J. Biomed. Sci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tardáguila, M.; González-Gugel, E.; Sánchez-Pacheco, A. Aurora Kinase B Activity Is Modulated by Thyroid Hormone during Transcriptional Activation of Pituitary Genes. Mol. Endocrinol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Stratford, A.L.; Fry, C.J.; Desilets, C.; Davies, A.H.; Cho, Y.Y.; Li, Y.; Dong, Z.; Berquin, I.M.; Roux, P.P.; Dunn, S.E. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Reipas, K.M.; Pambid, M.R.; Berns, R.; Stratford, A.L.; Fotovati, A.; Firmino, N.; Astanehe, A.; Hu, K.; Maxwell, C.; et al. YB-1 transforms human mammary epithelial cells through chromatin remodeling leading to the development of basal-like breast cancer. Stem Cells 2014. [Google Scholar] [CrossRef]
- Zhao, C.; Li, Y.; Zhang, W.; Zhao, D.; Ma, L.; Ma, P.; Yang, F.; Wang, Y.; Shu, Y.; Qiu, W. IL-17 induces NSCLC A549 cell proliferation via the upregulation of HMGA1, resulting in an increased cyclin D1 expression. Int. J. Oncol. 2018. [Google Scholar] [CrossRef]
- Harada, M.; Kotake, Y.; Ohhata, T.; Kitagawa, K.; Niida, H.; Matsuura, S.; Funai, K.; Sugimura, H.; Suda, T.; Kitagawa, M. YB-1 promotes transcription of cyclin D1 in human non-small-cell lung cancers. Genes Cells 2014. [Google Scholar] [CrossRef]
- Zanin, R.; Pegoraro, S.; Ros, G.; Ciani, Y.; Piazza, S.; Bossi, F.; Bulla, R.; Zennaro, C.; Tonon, F.; Lazarevic, D.; et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J. Exp. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Sgarra, R.; Maurizio, E.; Zammitti, S.; Lo Sardo, A.; Giancotti, V.; Manfioletti, G. Macroscopic differences in HMGA oncoproteins post-translational modifications: C-terminal phosphorylation of HMGA2 affects its DNA binding properties. J. Proteome Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Arnoldo, L.; Sgarra, R.; Chiefari, E.; Iiritano, S.; Arcidiacono, B.; Pegoraro, S.; Pellarin, I.; Brunetti, A.; Manfioletti, G. A novel mechanism of post-translational modulation of HMGA functions by the histone chaperone nucleophosmin. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penzo, C.; Arnoldo, L.; Pegoraro, S.; Petrosino, S.; Ros, G.; Zanin, R.; Wiśniewski, J.R.; Manfioletti, G.; Sgarra, R. HMGA1 Modulates Gene Transcription Sustaining a Tumor Signalling Pathway Acting on the Epigenetic Status of Triple-Negative Breast Cancer Cells. Cancers 2019, 11, 1105. https://doi.org/10.3390/cancers11081105
Penzo C, Arnoldo L, Pegoraro S, Petrosino S, Ros G, Zanin R, Wiśniewski JR, Manfioletti G, Sgarra R. HMGA1 Modulates Gene Transcription Sustaining a Tumor Signalling Pathway Acting on the Epigenetic Status of Triple-Negative Breast Cancer Cells. Cancers. 2019; 11(8):1105. https://doi.org/10.3390/cancers11081105
Chicago/Turabian StylePenzo, Carlotta, Laura Arnoldo, Silvia Pegoraro, Sara Petrosino, Gloria Ros, Rossella Zanin, Jacek R. Wiśniewski, Guidalberto Manfioletti, and Riccardo Sgarra. 2019. "HMGA1 Modulates Gene Transcription Sustaining a Tumor Signalling Pathway Acting on the Epigenetic Status of Triple-Negative Breast Cancer Cells" Cancers 11, no. 8: 1105. https://doi.org/10.3390/cancers11081105
APA StylePenzo, C., Arnoldo, L., Pegoraro, S., Petrosino, S., Ros, G., Zanin, R., Wiśniewski, J. R., Manfioletti, G., & Sgarra, R. (2019). HMGA1 Modulates Gene Transcription Sustaining a Tumor Signalling Pathway Acting on the Epigenetic Status of Triple-Negative Breast Cancer Cells. Cancers, 11(8), 1105. https://doi.org/10.3390/cancers11081105