Coagulation Factor Xa Promotes Solid Tumor Growth, Experimental Metastasis and Endothelial Cell Activation

 , , ,
, , ,  , , , and add
Show full author list
, , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Culture
2.2. Flow Cytometry
2.3. Regression Assay
2.4. In Vitro/In Vivo Permeability Assay
2.5. Immunofluorescence
2.6. Underflow and Static Adhesion Assay
2.7. Cell Migration and Invasion
2.8. Western Blot
2.9. Metastasis and Tumor Growth In Vivo Experiments
2.9.1. Microarray Analysis
2.9.2. Statistical Analysis
3. Results
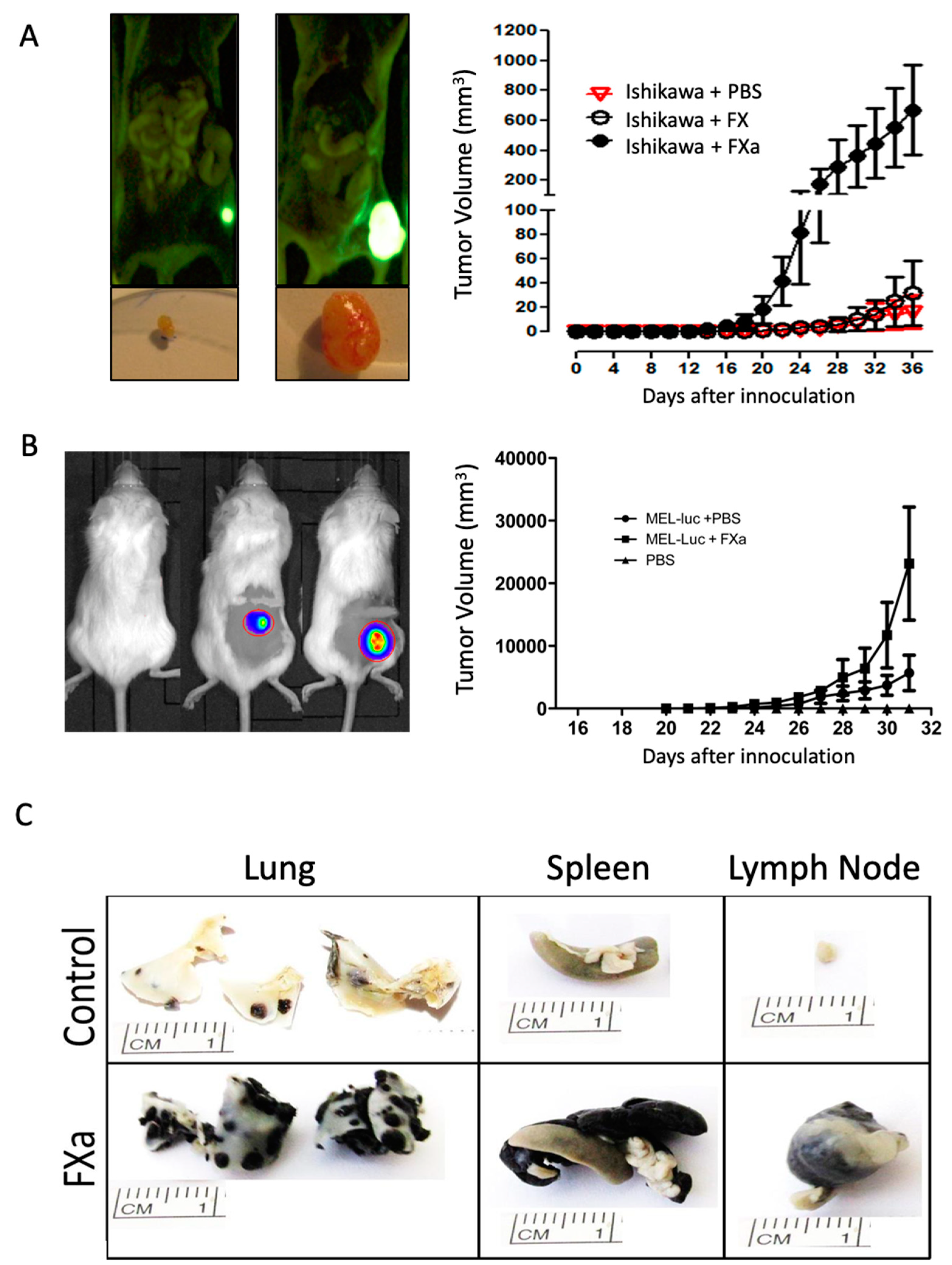
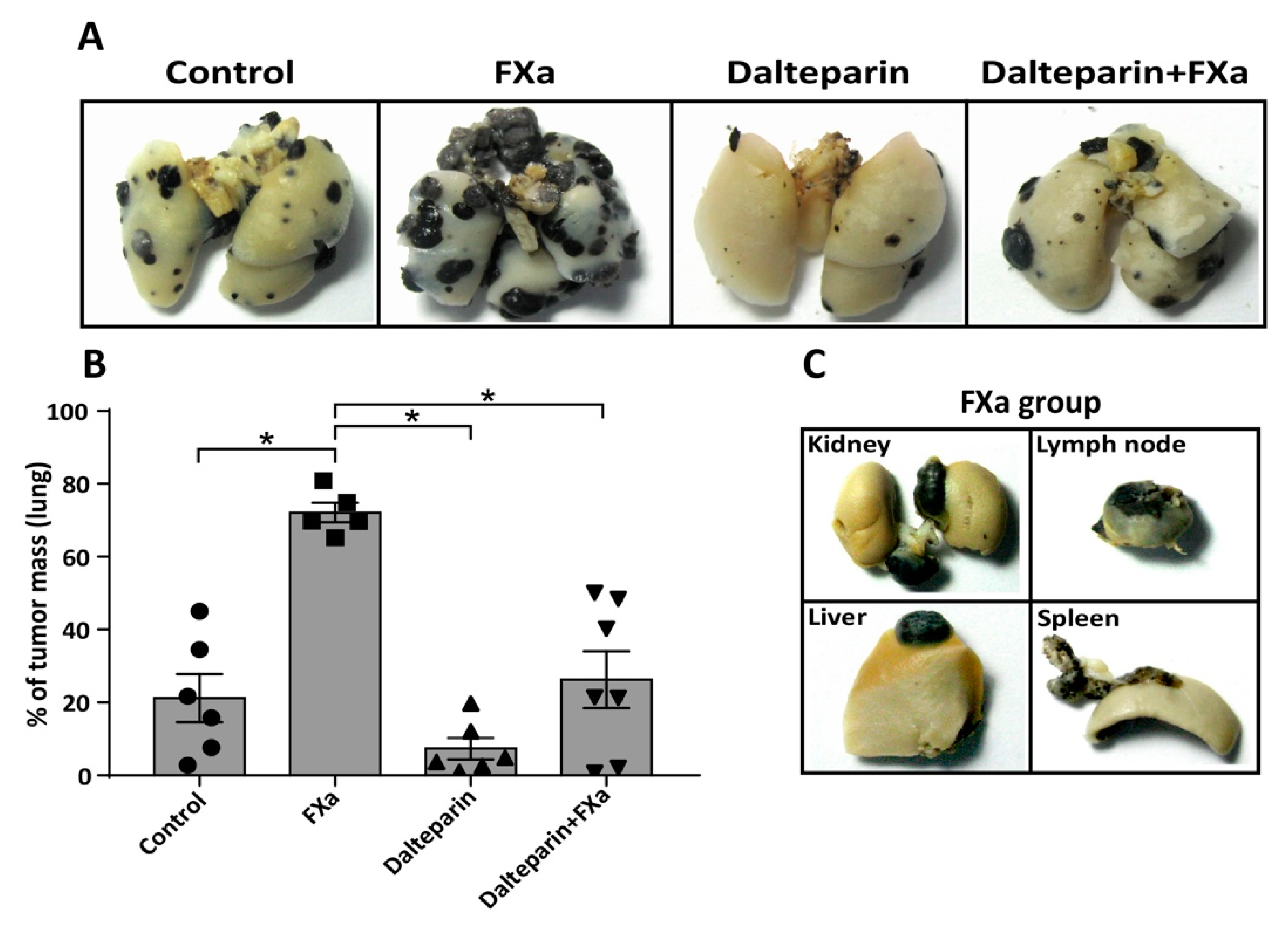
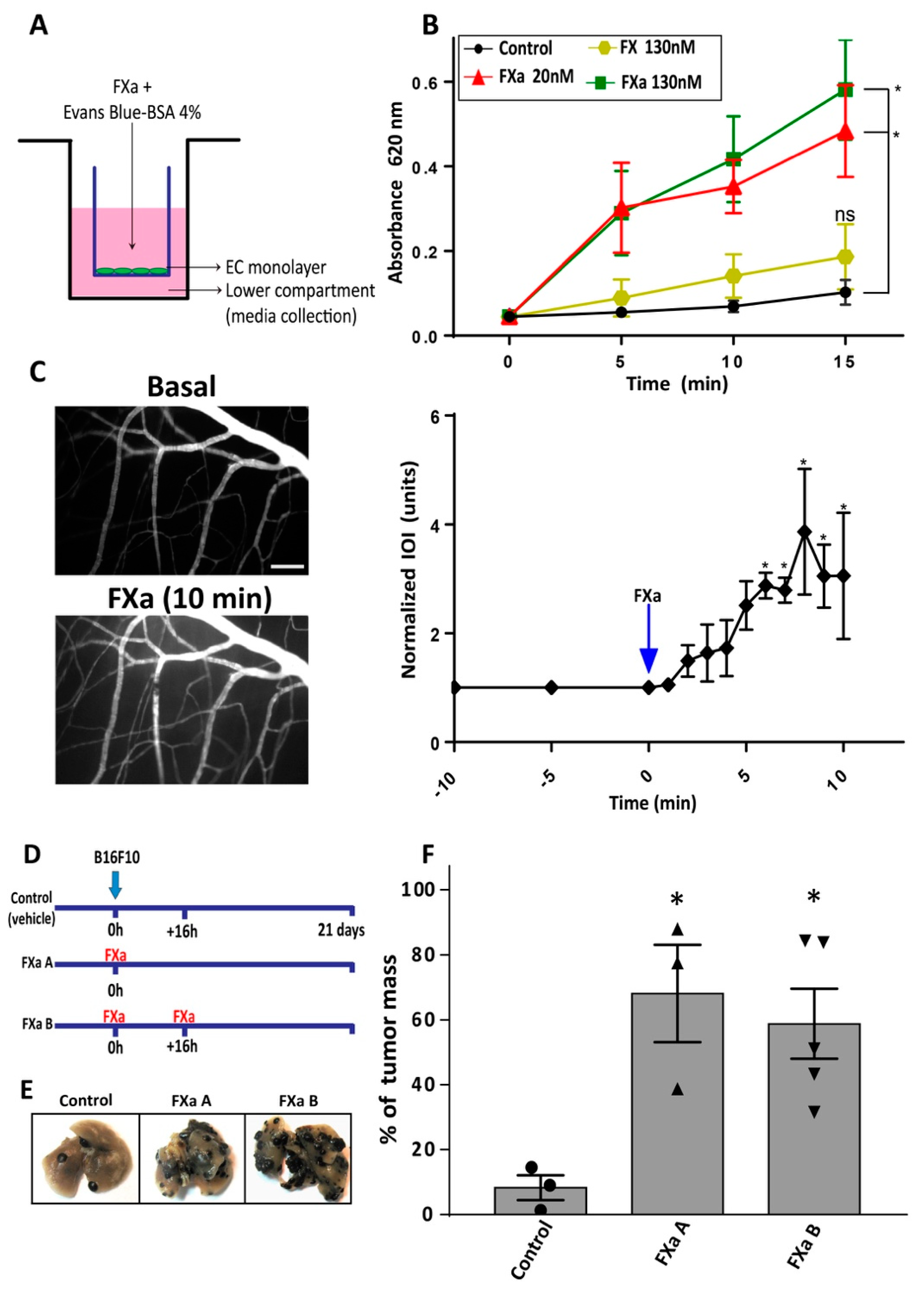
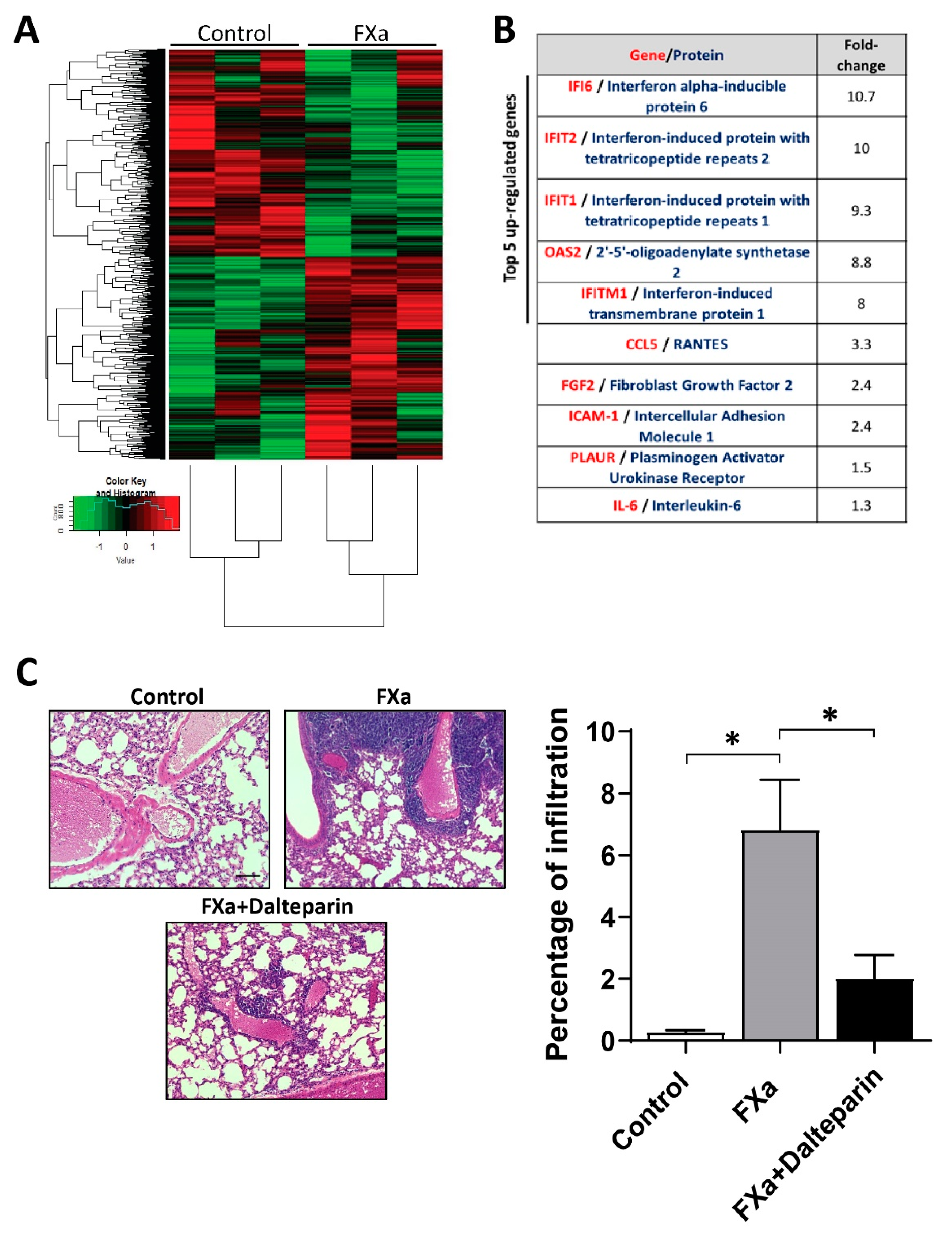
3.1. FXa Increased Metastasis In Vivo, An Effect Inhibited by Dalteparin Treatment
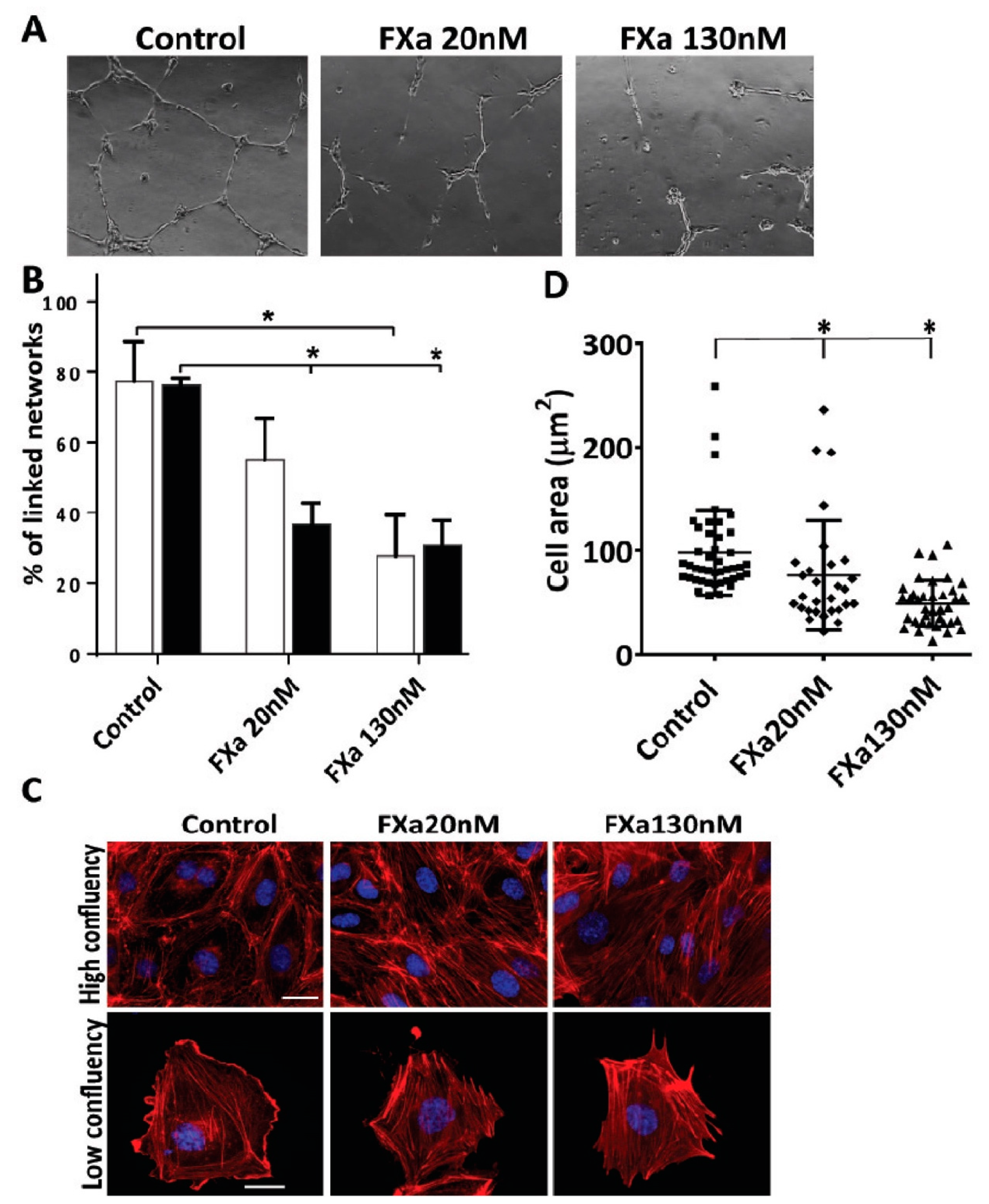
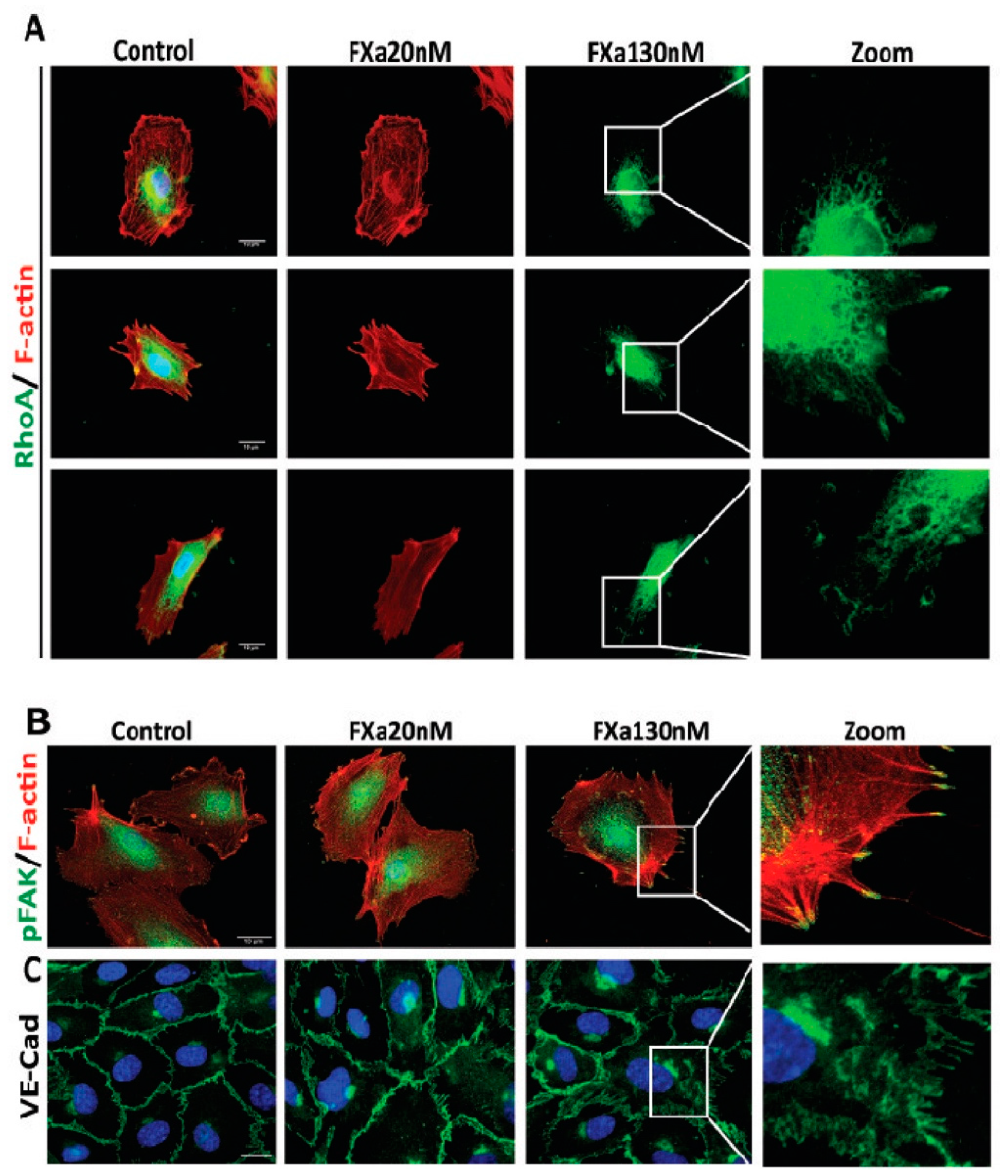
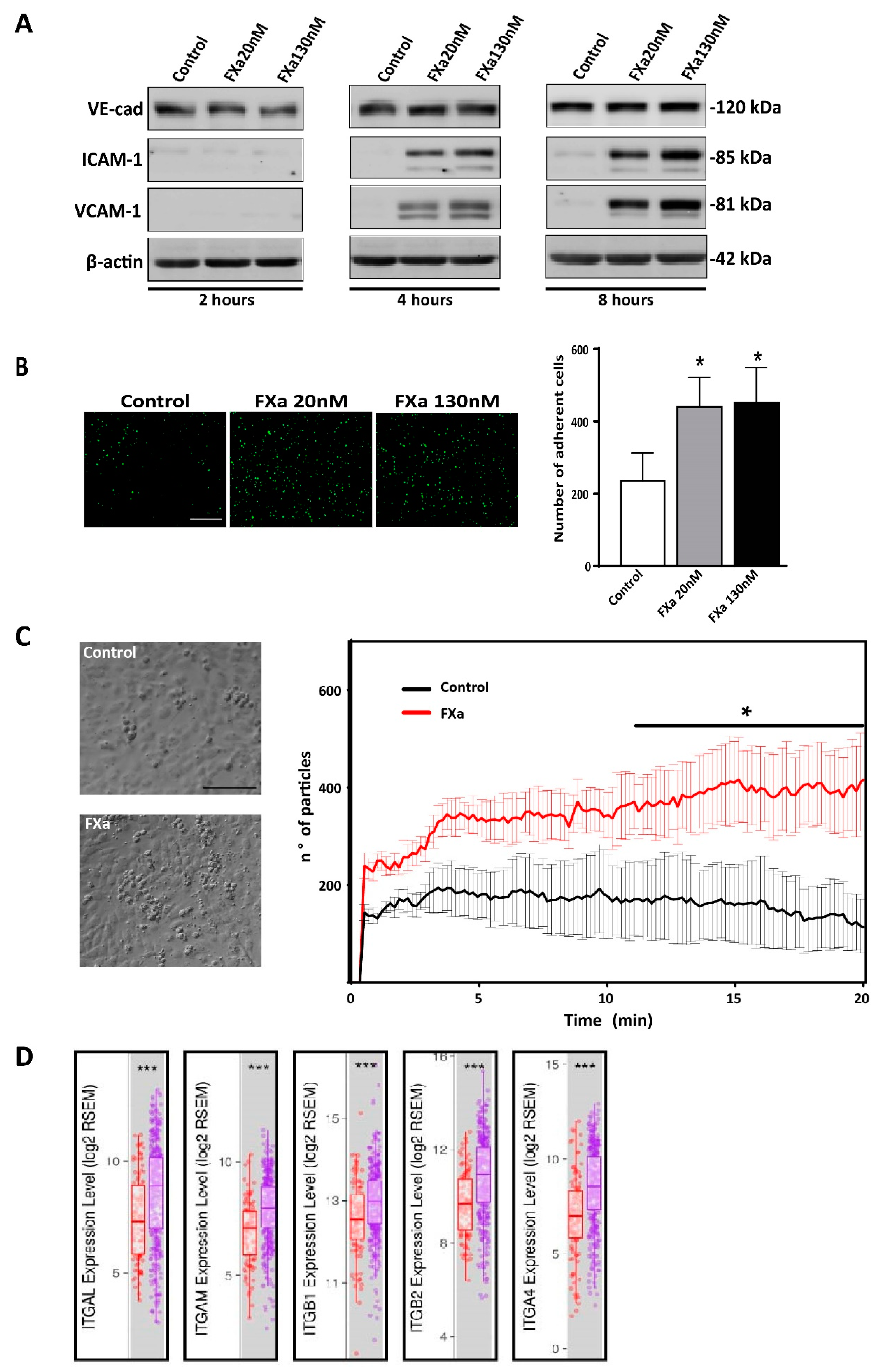
3.2. FXa Alters Endothelial Cell Morphology and Contractibility
3.3. FXa Induces Endothelial Hyperpermeability
3.4. FXa Changes mRNA Expression within Endothelial Cells
3.5. FXa Increases Cancer Cell Adhesion to the Endothelium
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Noble, J.; Pasi, J. Epidemiology and pathophysiology of cancer-associated thrombosis. Br. J. Cancer 2010, 102, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Elyamany, G.; Alzahrani, A.M.; Bukhary, E. Cancer-associated thrombosis: An overview. Clin. Med. Insights Oncol. 2014, 8, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M.; Vignoli, A. Coagulation and cancer: Biological and clinical aspects. J. Thromb. Haemost. 2013, 11, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Desch, A.; Gebhardt, C.; Utikal, J.; Schneider, S.W. D-dimers in malignant melanoma: Association with prognosis and dynamic variation in disease progress. Int. J. Cancer. 2017, 140, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Sparsa, A.; Durox, H.; Doffoel-Hantz, V.; Munyangango, E.M.; Bédane, C.; Cendras, J.; Gantois, C.; Boulinguez, S.; Bonnetblanc, J.M. High prevalence and risk factors of thromboembolism in stage IV melanoma. J. Eur. Acad. Dermatol. Venereol. 2011, 11, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Kasthuri, R.S.; Taubman, M.B.; Mackman, N. Role of tissue factor in cancer. J. Clin. Oncol. 2009, 11, 4834–4838. [Google Scholar] [CrossRef] [PubMed]
- Henriquez, S.; Calderon, C.; Quezada, M.; Oliva, B.; Bravo, M.L.; Aranda, E.; Kato, S.; Cuello, M.A.; Gutierrez, J.; Quest, A.F.; et al. Progesterone utilizes distinct membrane pools of tissue factor to increase coagulation and invasion and these effects are inhibited by TFPI. J. Cell Physiol. 2011, 226, 3278–3285. [Google Scholar] [CrossRef]
- Krupiczojc, M.A.; Scotton, C.J.; Chambers, R.C. Coagulation signalling following tissue injury: Focus on the role of factor Xa. Int. J. Biochem. Cell Biol. 2008, 18, 1228–1237. [Google Scholar] [CrossRef]
- Haddad, T.C.; Greeno, E.W. Chemotherapy-induced thrombosis. Thromb. Res. 2006, 118, 555–568. [Google Scholar] [CrossRef]
- Kirwan, C.C.; Mccollum, C.N.; Mcdowell, G.; Byrne, G.J. Investigation of Proposed Mechanisms of Chemotherapy-Induced Venous Thromboembolism: Endothelial Cell Activation and Procoagulant Release Due to Apoptosis. Clin. Appl. Thromb. 2015, 22, 420–427. [Google Scholar] [CrossRef]
- Laresche, C.; Pelletier, F.; Garnache-Ottou, F.; Lihoreau, T.; Biichlé, S.; Mourey, G.; Saas, P.; Humbert, P.; Seilles, E.; Aubin, F. Increased levels of circulating microparticles are associated with increased procoagulant activity in patients with cutaneous malignant melanoma. J. Investig. Dermatol. 2014, 134, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Zigler, M.; Kamiya, T.; Brantley, E.C.; Villares, G.J.; Bar-Eli, M. PAR-1 and thrombin: The ties that bind the microenvironment to melanoma metastasis. Cancer Res. 2011, 71, 6561–6566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, N.; Jin, K.; He, K.; Cao, J.; Teng, L. Protease-activated receptors in cancer: A systematic review. Oncol. Lett. 2011, 2, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Peppelenbosch, M.P.; Spek, C.A. Coagulation Factor Xa inhibits cancer cell migration via LIMK1-mediated cofilin inactivation. Thromb. Res. 2010, 5, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Senden, N.H.; Jeunhomme, T.M.; Heemskerk, J.W.; Wagenvoord, R.; van’t Veer, C.; Hemker, H.C.; Buurman, W.A. Factor Xa Induces Cytokine Production and Expression of Adhesion Molecules by Human Umbilical Vein Endothelial Cells. J. Immunol. 1998, 161, 4318–4324. [Google Scholar] [PubMed]
- Ebrahimi, S.; Rahmani, F.; Behnam-Rassouli, R.; Hoseinkhani, F.; Parizadeh, M.R.; Keramati, M.R.; Khazaie, M.; Avan, A.; Hassanian, S.M. Proinflammatory signaling functions of thrombin in cancer. J. Cell. Physiol. 2017, 232, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Läubli, H.; Borsig, L. Selectins as Mediators of Lung Metastasis. Cancer Microenviron. 2010, 3, 97–105. [Google Scholar] [CrossRef]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Ferjancic, Š.; Gil-Bernabé, A.M.; Hill, S.A.; Allen, P.D.; Richardson, P.; Sparey, T.; Savory, E.; McGuffog, J.; Muschel, R.J. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood 2013, 121, 3289–3297. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Dejana, E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 2007, 120, 1–6. [Google Scholar] [CrossRef]
- Reymond, N.; D’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- van Nieuw Amerongen, G.P.; van Delft, S.; Vermeer, M.A.; Collard, J.G.; van Hinsbergh, V.W. Activation of RhoA by Thrombin in Endothelial Hyperpermeability. Circ. Res. 2012, 87, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Gerotziafas, G.T.; Papageorgiou, C.; Hatmi, M.; Samama, M.M.; Elalamy, I. Clinical studies with anticoagulants to improve survival in cancer patients. Pathophysiol. Haemost. Thromb. 2008, 36, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.O.; Burns, S.; Noble, S.I.; Macbeth, F.R.; Cohen, D.; Maughan, T.S. FRAGMATIC: A randomised phase III clinical trial investigating the effect of fragmin® added to standard therapy in patients with lung cancer. BMC Cancer 2009, 9, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Altinbas, M.; Coskun, H.S.; Er, O.; Ozkan, M.; Eser, B.; Unal, A.; Cetin, M.; Soyuer, S. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J. Thromb. Haemost. 2004, 2, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Levine, M.N.; Kadziola, Z.; Lemoine, N.R.; Low, V.; Patel, H.K.; Rustin, G.; Thomas, M.; Quigley, M.; Williamson, R.C. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: The fragmin advanced malignancy outcome study (FAMOUS). J. Clin. Oncol. 2004, 22, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Albadawi, H.; Witting, A.A.; Pershad, Y.; Wallace, A.; Fleck, A.R.; Hoang, P.; Khademhosseini, A.; Oklu, R. Animal models of venous thrombosis. Cardiovasc. Diagn. Ther. 2017, 7, 197–206. [Google Scholar] [CrossRef]
- Kondreddy, V.; Wang, J.; Keshava, S.; Esmon, C.T.; Rao, L.V.M.; Pendurthi, U.R. Factor VIIa induces anti-inflammatory signaling via EPCR and PAR1. Blood 2018, 131, 2379–2392. [Google Scholar] [CrossRef] [PubMed]
- Torres-Estay, V.; Carreño, D.V.; Fuenzalida, P.; Watts, A.; San Francisco, I.F.; Montecinos, V.P.; Sotomayor, P.C.; Ebos, J.; Smith, G.J.; Godoy, A.S. Androgens modulate male-derived endothelial cell homeostasis using androgen receptor-dependent and receptor-independent mechanisms. Angiogenesis 2016, 16, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Oyarce, C.; Cruz-Gomez, S.; Galvez-Cancino, F.; Vargas, P.; Moreau, H.D.; Diaz-Valdivia, N.; Diaz, J.; Salazar-Onfray, F.A.; Pacheco, R.; Lennon-Dumenil, A.M.; et al. Caveolin-1 expression increases upon maturation in dendritic cells and promotes their migration to lymph nodes thereby favoring the induction of CD8+ T cell responses. Front. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.E.; Rhoades, R.A.; Garcia, J.G. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J. Appl. Physiol. 1992, 72, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Marín, N.; Zamorano, P.; Carrasco, R.; Mujica, P.; González, F.G.; Quezada, C.; Meininger, C.J.; Boric, M.P.; Durán, W.N.; Sánchez, F.A. S-Nitrosation of β-catenin and p120 catenin: A novel regulatory mechanism in endothelial hyperpermeability. Circ. Res. 2012, 111, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Durán, W.N.; Seyama, A.; Yoshimura, K.; González, D.R.; Jara, P.I.; Figueroa, X.F.; Borić, M.P. Stimulation of NO production and of eNOS phosphorylation in the microcirculation in vivo. Microvasc. Res. 2000, 60, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Bustos, M.A.; Lucchesi, O.; Ruete, M.C.; Mayorga, L.S.; Tomes, C.N. Rab27 and Rab3 sequentially regulate human sperm dense-core granule exocytosis. Proc. Natl. Acad. Sci. USA 2012, 109, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Erices, R.; Cubillos, S.; Aravena, R.; Santoro, F.; Marquez, M.; Orellana, R.; Ramírez, C.; González, P.; Fuenzalida, P.; Bravo, M.L.; et al. Diabetic concentrations of metformin inhibit platelet-mediated ovarian cancer cell progression. Oncotarget 2017, 8, 20865–20880. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Bunce, M.W.; Toso, R.; Camire, R.M. Zymogen-like factor Xa variants restore thrombin generation and effectively bypass the intrinsic pathway in vitro. Blood 2011, 117, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.R. Heparin-binding exosite of factor Xa. Trends Cardiovasc. Med. 2000, 10, 333–338. [Google Scholar] [CrossRef]
- Buijs, J.T.; Laghmani, E.H.; van den Akker, R.F.P.; Tieken, C.; Vletter, E.M.; van der Molen, K.M.; Crooijmans, J.J.; Kroone, C.; Le Dévédec, S.E.; van der Pluijm, G.; et al. The Direct Oral Anticoagulants Rivaroxaban and Dabigatran do not Inhibit Orthotopic Growth and Metastasis of Human Breast Cancer in Mice. J. Thromb. Haemost. 2019, 17, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Chan, J.S.; Lee, C.Y.; Leu, H.B.; Huang, P.H.; Chen, J.S.; Lin, S.J.; Chen, J.W. Rivaroxaban, a factor Xa inhibitor, improves neovascularization in the ischemic hindlimb of streptozotocin-induced diabetic mice. Cardiovasc. Diabetol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Feistritzer, C.; Lenta, R.; Riewald, M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: Role in factor Xa signaling. J. Thromb. Haemost. 2005, 3, 2798–2805. [Google Scholar] [CrossRef]
- Schuepbach, R.A.; Riewald, M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C. receptor. J. Thromb. Haemost. 2010, 8, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Artim-Esen, B.; Smoktunowicz, N.; McDonnell, T.; Ripoll, V.M.; Pericleous, C.; MacKie, I.; Robinson, E.; Isenberg, D.; Rahman, A.; Ioannou, Y.; et al. Factor Xa Mediates Calcium Flux in Endothelial Cells and is Potentiated by Igg from Patients with Lupus and/or Antiphospholipid Syndrome. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Lange, S.; Gonzalez, I.; Pinto, M.P.; Arce, M.; Valenzuela, R.; Aranda, E.; Elliot, M.; Alvarez, M.; Henriquez, S.; Velasquez, E.V.; et al. Independent Anti-Angiogenic Capacities of Coagulation Factors, X. and Xa. J. Cell Physiol. 2014, 229, 1673–1680. [Google Scholar] [CrossRef]
- Benelhaj, N.E.; Maraveyas, A.; Featherby, S.; Collier, M.E.W.; Johnson, M.J.; Ettelaie, C. Alteration in endothelial permeability occurs in response to the activation of PAR2 by factor Xa but not directly by the TF-factor VIIa complex. Thromb. Res. 2019, 175, 13–20. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Tang, Y.M.; Cao, Q.Y.; Guo, X.Y.; Dong, S.H.; Duan, J.A.; Wu, Q.N.; Liang, Q.L. Inhibition of p38 and ERK1/2 pathways by Sparstolonin B suppresses inflammation-induced melanoma metastasis. Biomed. Pharmacother. 2018, 98, 382–389. [Google Scholar] [CrossRef]
- Natoni, A.; Macauley, M.S.; O’Dwyer, M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Dass, K.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Sarkar, F.H. Evolving role of uPA/uPAR system in human cancers. Cancer Treat. Rev. 2008, 34, 122–136. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arce, M.; Pinto, M.P.; Galleguillos, M.; Muñoz, C.; Lange, S.; Ramirez, C.; Erices, R.; Gonzalez, P.; Velasquez, E.; Tempio, F.; et al. Coagulation Factor Xa Promotes Solid Tumor Growth, Experimental Metastasis and Endothelial Cell Activation. Cancers 2019, 11, 1103. https://doi.org/10.3390/cancers11081103
Arce M, Pinto MP, Galleguillos M, Muñoz C, Lange S, Ramirez C, Erices R, Gonzalez P, Velasquez E, Tempio F, et al. Coagulation Factor Xa Promotes Solid Tumor Growth, Experimental Metastasis and Endothelial Cell Activation. Cancers. 2019; 11(8):1103. https://doi.org/10.3390/cancers11081103
Chicago/Turabian StyleArce, Maximiliano, Mauricio P. Pinto, Macarena Galleguillos, Catalina Muñoz, Soledad Lange, Carolina Ramirez, Rafaela Erices, Pamela Gonzalez, Ethel Velasquez, Fabián Tempio, and et al. 2019. "Coagulation Factor Xa Promotes Solid Tumor Growth, Experimental Metastasis and Endothelial Cell Activation" Cancers 11, no. 8: 1103. https://doi.org/10.3390/cancers11081103
APA StyleArce, M., Pinto, M. P., Galleguillos, M., Muñoz, C., Lange, S., Ramirez, C., Erices, R., Gonzalez, P., Velasquez, E., Tempio, F., Lopez, M. N., Salazar-Onfray, F., Cautivo, K., Kalergis, A. M., Cruz, S., Lladser, Á., Lobos-González, L., Valenzuela, G., Olivares, N., ... Owen, G. I. (2019). Coagulation Factor Xa Promotes Solid Tumor Growth, Experimental Metastasis and Endothelial Cell Activation. Cancers, 11(8), 1103. https://doi.org/10.3390/cancers11081103