Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides



Abstract
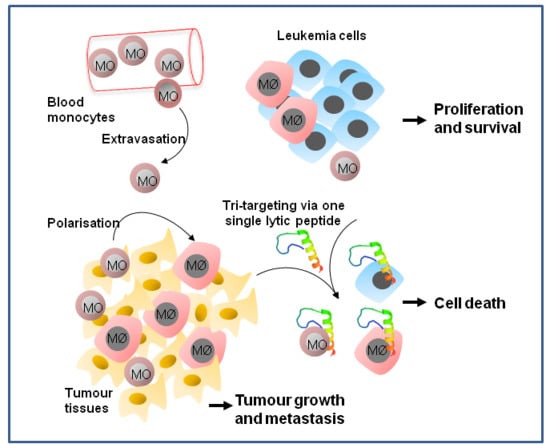
{kind=link}
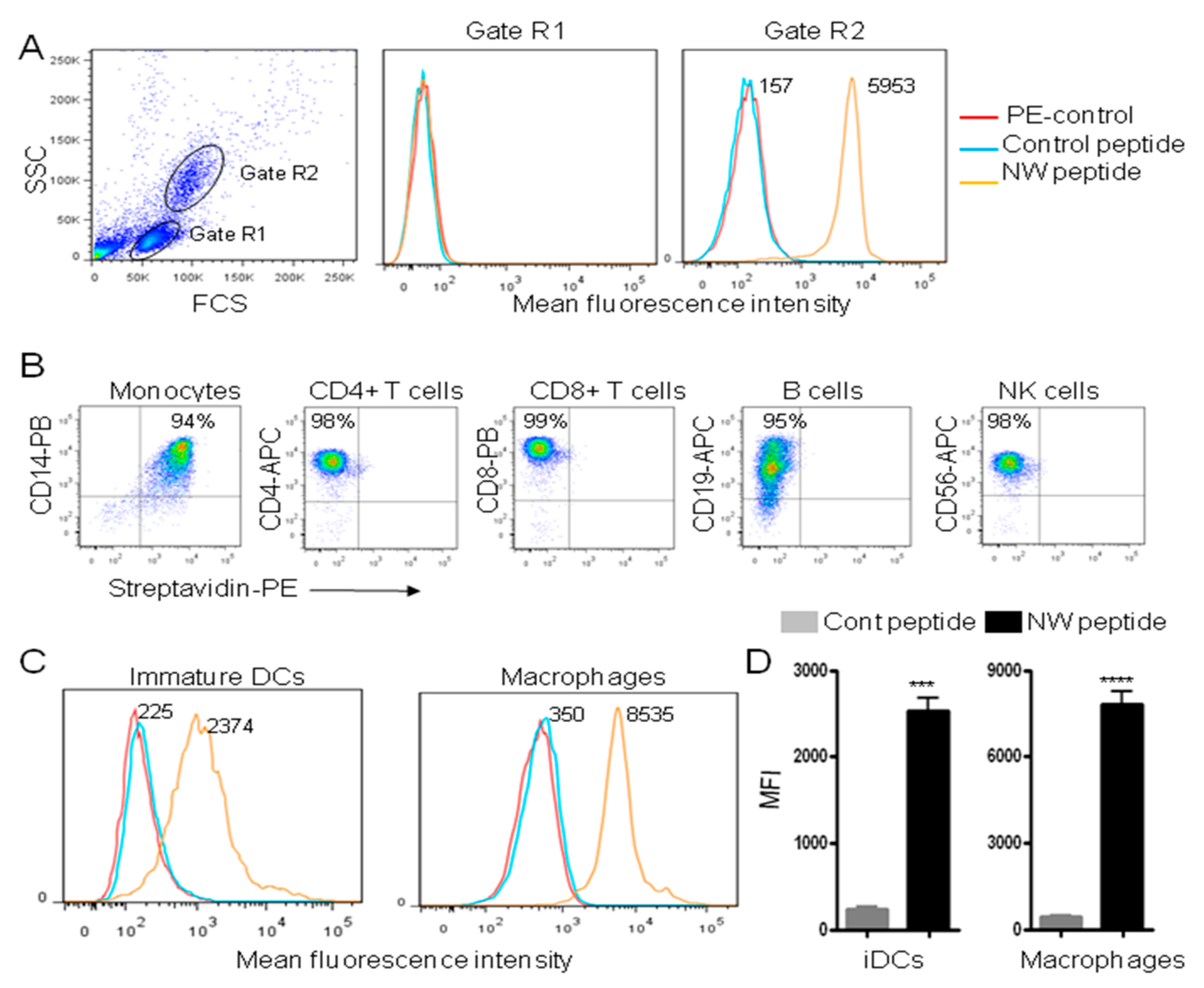{kind=link}
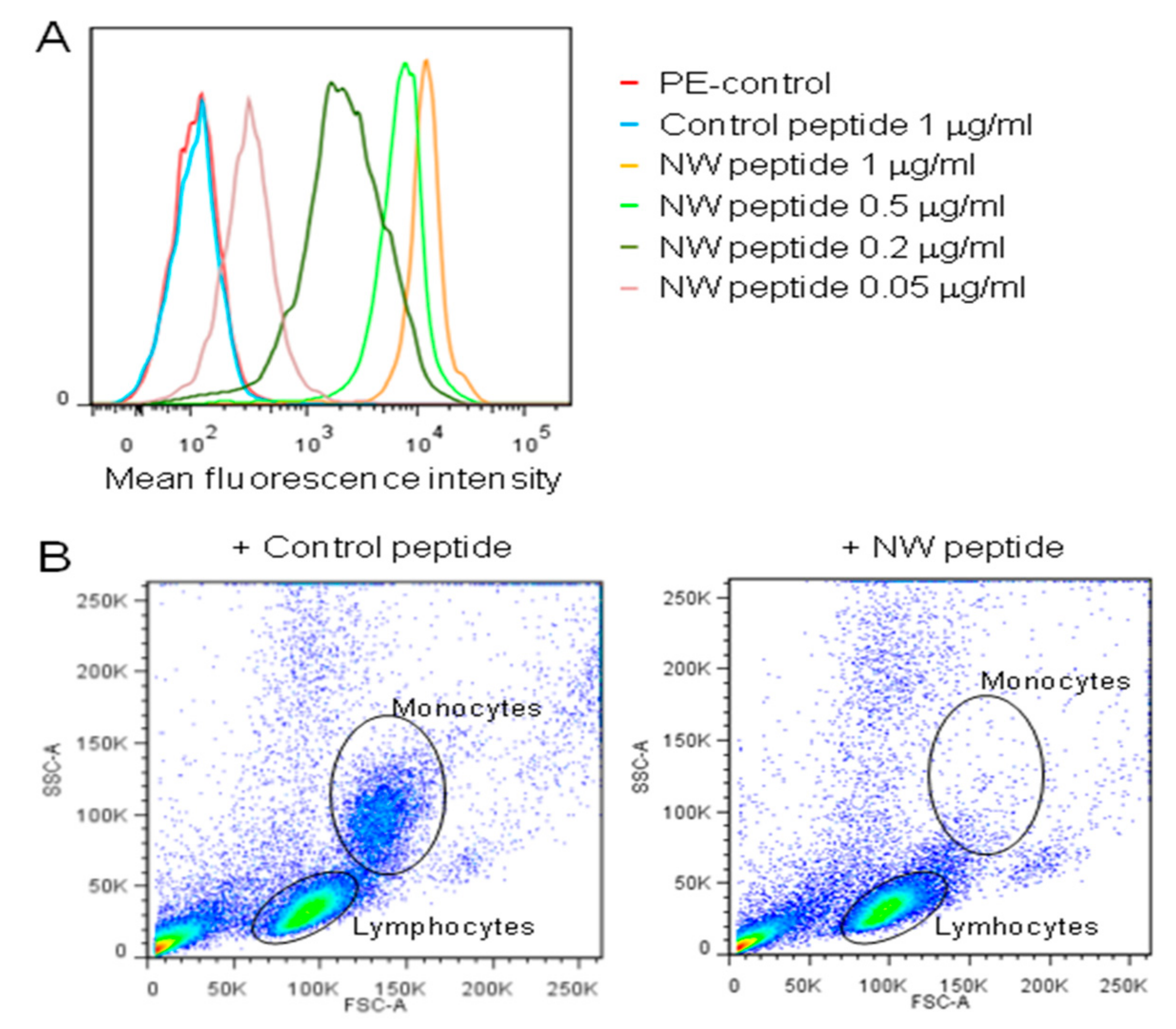{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
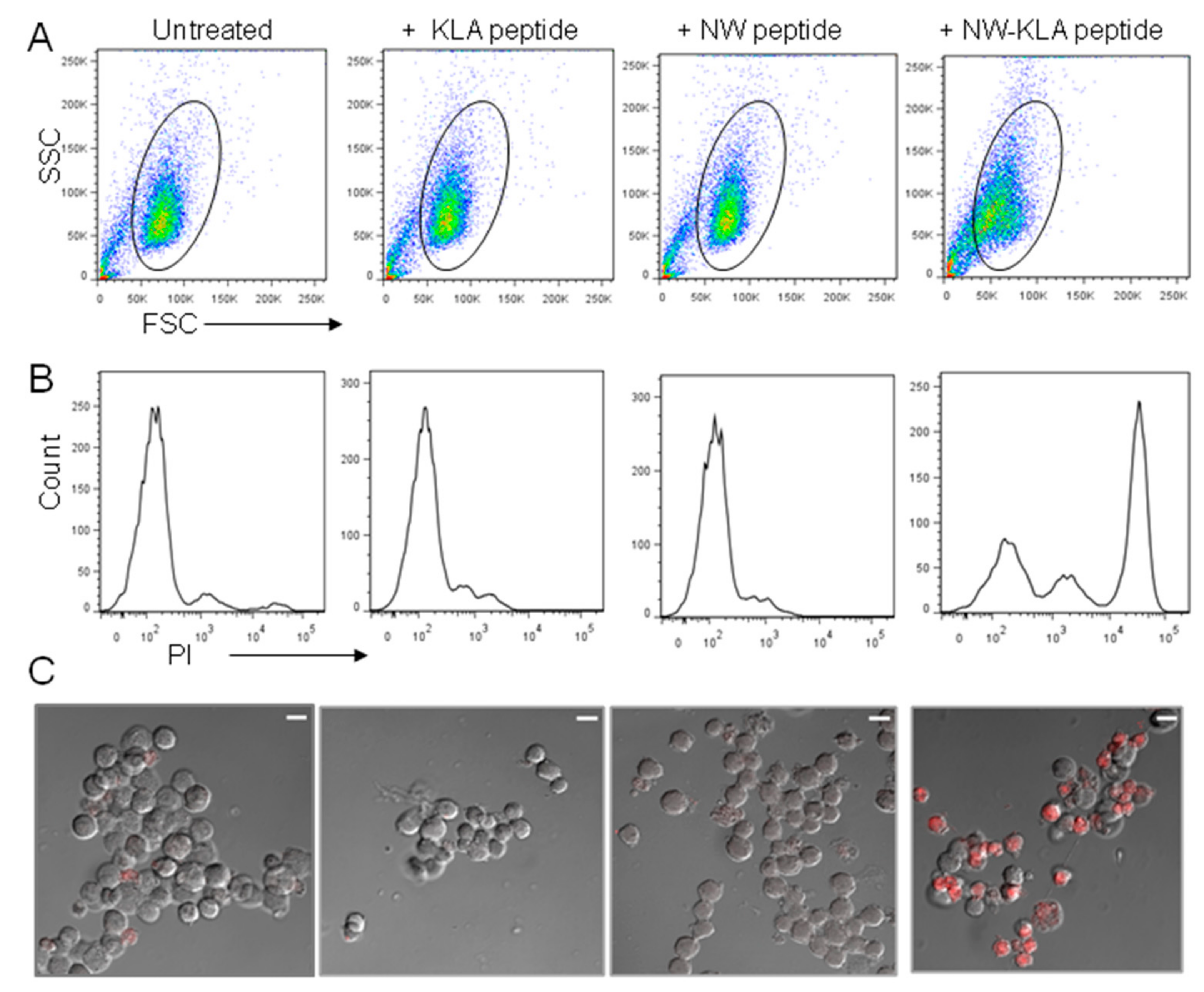{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Peptides
- NW peptide: NWYLPWLGTNDW-NH2
- NW peptide-biotin: NWYLPWLGTNDW-GGK-biotin
- Control peptide: MEWSLEKGYTIK-GGK-biotin
- KLA peptide: KLAKLAKKLAKLAK-NH2
- KLL peptide: KLLLKLLKKLLKLLKKK-NH2
- QLG peptide: QLGKKKHRRRPSKKRHW-NH2
- NW-KLA: NWYLPWLGTNDWGGGKLAKLAKKLAKLAK-NH2
- NW-KLL: NWYLPWLGTNDWGGGKLLLKLLKKLLKLLKKK-NH2
- NW-QLG: NWYLPWLGTNDWGGGQLGKKKHRRRPSKKRHW-NH2
2.2. Antibodies and Cytokines
2.3. Cell Lines and Peripheral Blood Mononuclear Cells
2.4. Primary Leukemia Cells
2.5. Generation of Immature Human Dendritic Cells
2.6. Generation of M1 and M2 Macrophages
2.7. Flow Cytometry
2.8. Depletion of Monocytes and Blast Cells from Peripheral Blood Mononuclear Cells
2.9. Cell Viability Assays
2.10. Apoptosis Assay
2.11. Uptake of the NW-Peptide Streptavidin-PE-Complexes by Macrophages
2.12. Statistical Analysis
3. Results
3.1. The NW Peptide Displays Strong Binding to Human Monocytes
3.2. Specific Killing of Monocytes by a Lytic Hybrid Peptide
3.3. Effects of the Fusion Lytic Peptides on M1 and M2 Macrophages
3.4. Effects of the Fusion Lytic Peptides on Leukemia Cell Lines
3.5. Effects of the Lytic Peptides on Primary Leukemia Cells
3.6. Depletion of Blood Blast Cells
3.7. Effects of the NW-KLA Peptide on Primary Mammary Epithelial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Recht, L.; Strober, S. The promise of targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-associated macrophages in ovarian cancer as targets for therapy. Cancers 2018, 10, 366. [Google Scholar] [CrossRef]
- Davignon, J.L.; Hayder, M.; Baron, M.; Boyer, J.F.; Constantin, A.; Apparailly, F.; Poupot, R.; Cantagrel, A. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Reumatology 2013, 52, 590–598. [Google Scholar] [CrossRef]
- Roberts, C.A.; Dickinson, A.K.; Taams, L.S. The Interplay Between Monocytes/Macrophages and CD4+ T Cell Subsets in Rheumatoid Arthritis. Front Immunol. 2015, 6, 571. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.K.; Li, Y.; Dang, Q.; Yang, F. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and, their heterogeneity and plasticity role in RA pathogenesis. Int. Immunopharmacol. 2018, 65, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Balhara, J.; Gounni, A.S. The alveolar macrophages in asthma: A double-edged sword. Mucosal Immunol. 2012, 5, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Keam, S.J. Trabectedin: A review of its use in soft tissue sarcoma and ovarian cancer. Drugs 2010, 70, 355–376. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Thompson, K.; Ebetino, F.H.; Rogers, M.J.; Coxon, F.P. Bisphosphonates: Molecular mechanisms of action and effects on bone cells, monocytes and macrophages. Curr. Pharm. Des. 2010, 16, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Zins, K.; Sioud, M.; Aharinejad, S.; Lucas, T.; Abraham, D. Modulating the tumor microenvironment with RNA interference as a cancer treatment strategy. Methods Mol. Biol. 2015, 1218, 143–161. [Google Scholar]
- Papo, N.; Shai, Y. Host defence peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 2005, 62, 785–790. [Google Scholar] [CrossRef]
- Leuschner, C.; Hansel, W. Membrane disrupting lytic peptides for cancer treatments. Curr. Pharm. Des. 2004, 10, 2299–2310. [Google Scholar] [CrossRef]
- Barua, S.; Linton, R.S.; Gamboa, J.; Banerjee, I.; Yarmush, M.L.; Rege, K. Lytic peptide-mediated sensitization of TRAIL-resistant prostate cancer cells to death receptor agonists. Cancer Lett. 2010, 293, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Mobergslien, A. Selective killing of cancer cells by peptide-targeted elivery of an anti-microbial peptide. Biochem. Pharm. 2012, 84, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Rege, K.; Patel, S.J.; Megeed, Z.; Yarmush, M.L. Amphipathic peptide-based fusion peptides and immunoconjugates for the targeted ablation of prostate cancer cells. Cancer Res. 2007, 67, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M.; Skorstad, G.; Mobergslien, A.; Sæbøe-Larssen, S. A novel peptide carrier for efficient targeting of antigens and nucleic acids to dendritic cells. FASEB J. 2013, 27, 3272–3283. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Phage Display Libraries: From binders to targeted drug delivery and human therapeutics. Mol. Biotechnol. 2019, 61, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Galán, A.; Comor, L.; Horvatić, A.; Kuleš, J.; Guillemin, N.; Mrljak, V.; Bhide, M. Library-based display technologies: Where do we stand? Mol. Biosyst. 2016, 12, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Gray, D.H. Isolation of thymic epithelial cells and analysis by flow cytometry. Curr. Protoc. Immunol. 2014, 107, 3–26. [Google Scholar] [PubMed]
- Neo, S.H.; Lew, Q.J.; Koh, S.M.; Zheng, L.; Bi, X.; Chao, S.H. Use of a novel cytotoxic HEXIM1 peptide in the directed breast cancer therapy. Oncotarget 2015, 7, 5483–5494. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Arakawa, S.; Nakanomyo, I.; Kudo-Sakamoto, Y.; Akazawa, H.; Komuro, I.; Shimizu, S. Identification of a novel compound that inhibits both mitochondria-mediated necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2015, 467, 1006–1011. [Google Scholar] [CrossRef]
- Lecoeur, H.; Prévost, M.C.; Gougeon, M.L. Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: A reconsideration of the specificity of the annexin V/propidium iodide assay. Cytometry 2001, 44, 65–72. [Google Scholar] [CrossRef]
- Zargarian, S.; Shlomovitz, I.; Erlich, Z.; Hourizadeh, A.; Ofir-Birin, Y.; Croker, B.A.; Regev-Rudzki, N.; Edry-Botzer, L.; Gerlic, M. Phosphatidylserine externalization, "necroptotic bodies" release, and phagocytosis during necroptosis. PLoS Biol. 2017, 15, e2002711. [Google Scholar] [CrossRef] [PubMed]
- Rey-Giraud, F.; Hafner, M.; Ries, C.H. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLoS ONE 2012, 7, e42656. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Scia, A.; Mantovani, A.; Locati, M. Macrohagae activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Horibe, T.; Haramoto, M.; Yano, Y.; Ohara, K.; Nakajima, O.; Matsuzaki, K.; Kawakami, K. A novel hybrid peptide targeting EGFR-expressing cancers. Eur. J. Cancer 2011, 47, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of relapsed/refractory acute myeloid leukemia. Curr. Treat. Opt. Oncol. 2017, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Guinn, B.A.; Mohamedali, A.; Thomas, N.S.B.; Mills, K.I. Immunotherapy of myeloid leukaemia. Cancer Immunol. Immunother. 2007, 56, 943–957. [Google Scholar] [CrossRef]
- Canals, C.; Torrico, C.; Picón, M.; Amill, B.; Cancelas, J.A.; Fraga, G.; Badell, I.; Cubells, J.; Olivé, T.; Ortega, J.; et al. Immunomagnetic bone marrow purging in children with acute lymphoblastic leukemia. J. Hematother. 1997, 6, 261–268. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeoloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Al-Matary, Y.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lames, R.F.; Thivakaran, A.; Schutte, J.; Köster, R.; Lennartz, K.; Schroeder, T.; et al. Acute meyeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef]
- Van Attekum, M.; Terpstra, S.; Reinen, E.; Kater, A.; Eldering, E. Macrophage-mediated chronic lymphocytic leukemia cell survival is independent of APRIL signaling. Cell Death Discov. 2016, 2, 16020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galletti, G.; Scielzo, C.; Barbaglio, F.; Rodriguez, T.V.; Riba, M.; Lazarevic, D.; Cittaro, D.; Simonetti, G.; Ranghetti, P.; Scarfò, L.; et al. Targeting macrophages sensitizes chronic lymphocytic leukemia to apoptosis and inhbit disease progression. Cell Rep. 1016, 14, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schäfer, R.; Stanley, E.R.; Abraham, D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Rowe, J.M.; Tallman, M.S. How I treat acute myeloid leukemia. Blood 2010, 116, 3147–3156. [Google Scholar] [CrossRef]
- Pandey, K.B.; Srivastava, S.; Singh, M.; Ghosh, J.K. Inducing toxicity by introducing a leucine-Zipper-like motif in from antimicrobial peptides, magainin 2. Biochem. J. 2011, 436, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; De Benedittis, C.; Mancini, M.; Martinelli, G. Best practices in chronic myeloid leukemia monitoring and managment. Oncologist 2016, 21, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Saubele, S.; Silver, R.T. Management of chronic myeloid leukemia in blast crisis. Ann. Hematol. 2015, 94, S159–S165. [Google Scholar]
- Pham, L.V.; Pogue, E.; Ford, R.J. The role of macrophage/B cell interactions in the pathophysiology of B cell lymphomas. Front. Oncol. 2018, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Hou, Y.; Sun, W.; Yu, J.; Liu, X.; Niu, Y.; Lu, J.J.; Chen, X. Natural products to prevent drug resistance in cancer chemotherapy: A review. Ann. N. Y. Acad. Sci. 2017, 1401, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.J.; Cooper, M.S.; Anderson, R.J.; Orchard, K.H.; Hale, G.; North, J.M.; Ganeshaguru, K.; Steele, A.J.; Mehta, A.B.; Lowdell, M.W.; et al. Selective apoptotic killing of malignant hemopoietic cells by antibody-targeted delivery of an amphipathic peptide. Cancer Res. 2005, 65, 2373–2377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aronson, M.R.; Simonson, A.W.; Orchard, L.M.; Llinás, M.; Medina, S.H. Lipopeptisomes: Anticancer peptide-assembled particles for fusolytic oncotherapy. Acta Biomater. 2018, 80, 269–277. [Google Scholar] [CrossRef]
- Gonzalez-Horta, A.; Matamoros-Acosta, A.; Chavez-Montes, A.; Castro-Rios, R.; Lara-Arias, J. Biodegradable nanoparticles loaded with tetrameric melittin: Preparation and membrane disruption evaluation. Gen. Physiol. Biophys. 2017, 36, 373–381. [Google Scholar] [CrossRef]
- Dybwad, A.; Førre, O.; Natvig, J.B.; Sioud, M. Structural characterization of peptides that bind synovial fluid antibodies from RA patients: A novel strategy for identification of disease-related epitopes using a random peptide library. Clin. Immunol. Immunopathol. 1995, 75, 45–50. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sioud, M.; Pettersen, S.; Ailte, I.; Fløisand, Y. Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides. Cancers 2019, 11, 1088. https://doi.org/10.3390/cancers11081088
Sioud M, Pettersen S, Ailte I, Fløisand Y. Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides. Cancers. 2019; 11(8):1088. https://doi.org/10.3390/cancers11081088
Chicago/Turabian StyleSioud, Mouldy, Solveig Pettersen, Ieva Ailte, and Yngvar Fløisand. 2019. "Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides" Cancers 11, no. 8: 1088. https://doi.org/10.3390/cancers11081088
APA StyleSioud, M., Pettersen, S., Ailte, I., & Fløisand, Y. (2019). Targeted Killing of Monocytes/Macrophages and Myeloid Leukemia Cells with Pro-Apoptotic Peptides. Cancers, 11(8), 1088. https://doi.org/10.3390/cancers11081088