Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
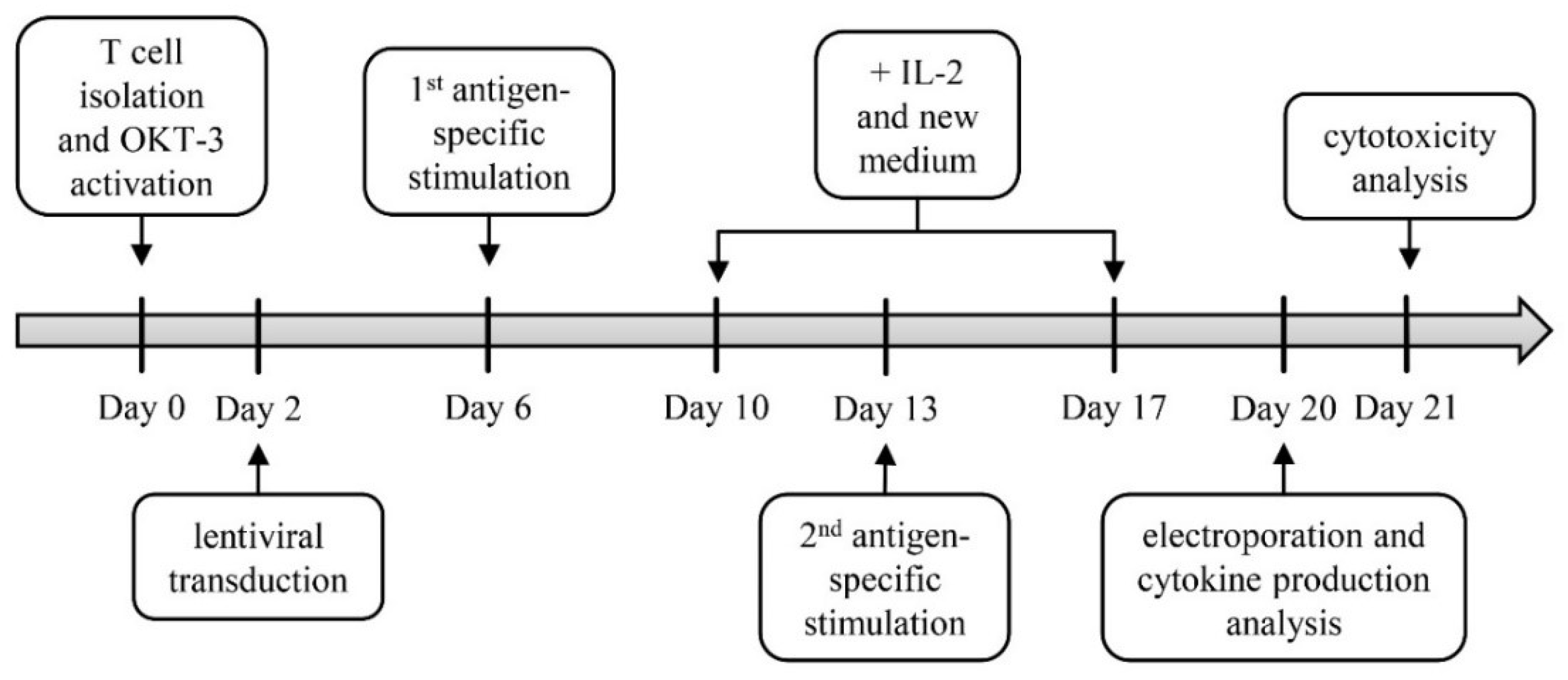
2.1. Generation of TETARs
2.2. Gp100-Specific TCR T Cells Can Be Efficiently Transfected with a CSPG4-Specific CAR Using mRNA Electroporation
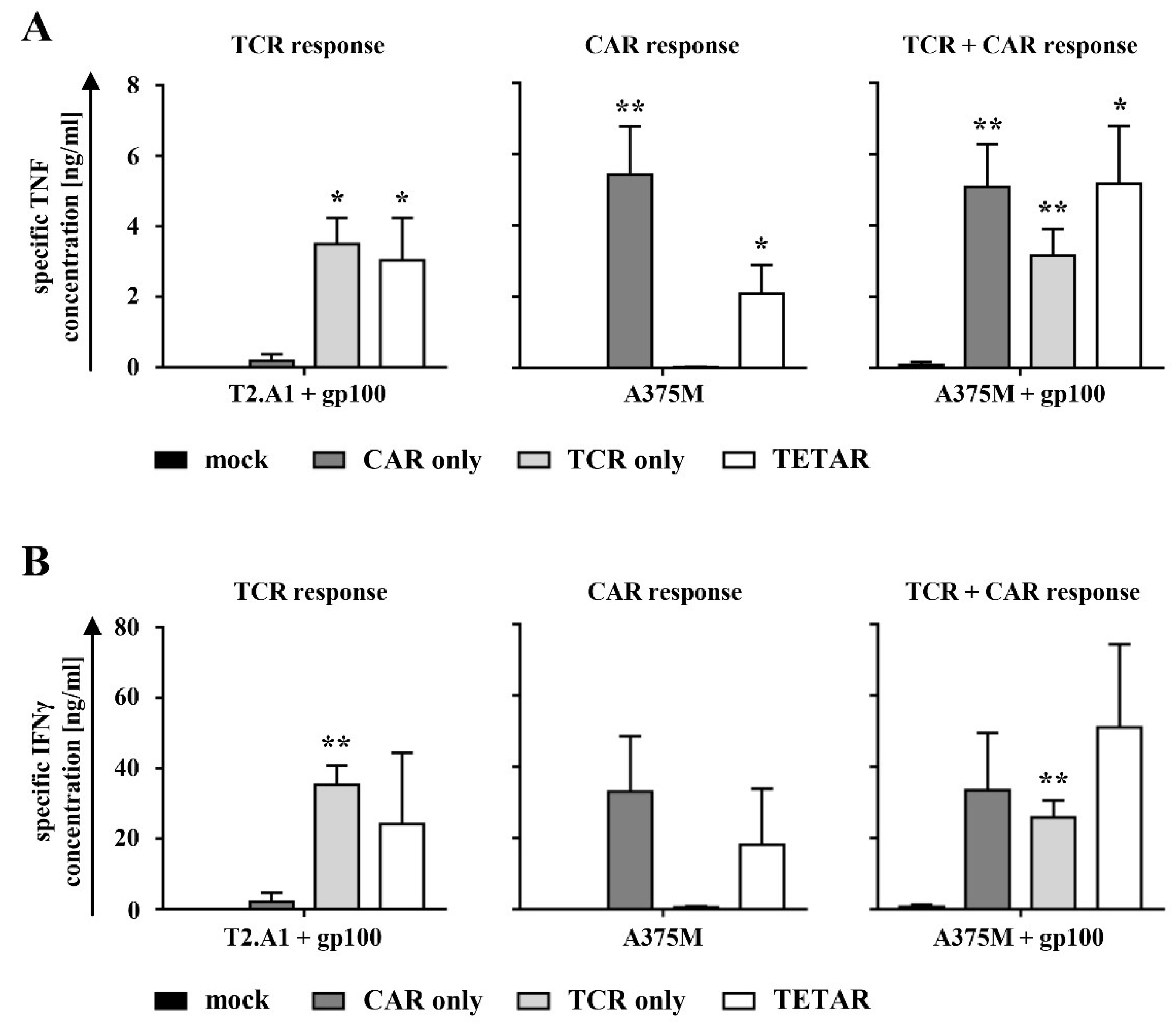
2.3. TETARs Antigen-Specifically Secrete Cytokines
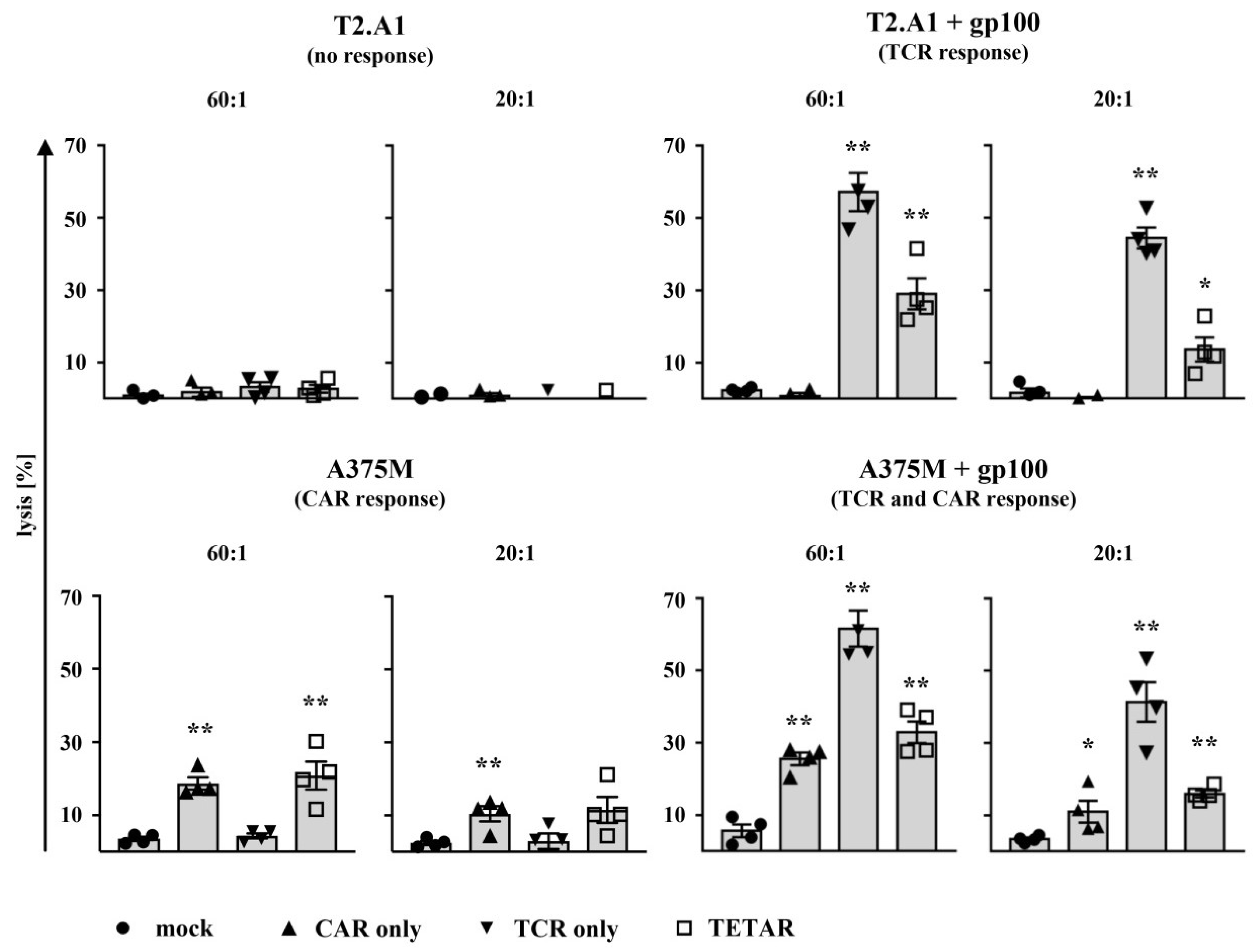
2.4. TETARs Antigen-Specifically Eliminate Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Lentiviral Transduction of T Cells
4.3. RNA Production and Transfection
4.4. Receptor Expression Analysis of Engineered T Cells
4.5. Cytokine Secretion Analysis of Engineered T Cells
4.6. Cytotoxicity Analysis of Engineered T Cells
4.7. Figure Preparation and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor- and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients with Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Olson, B.M.; McNeel, D.G. Antigen loss and tumor-mediated immunosuppression facilitate tumor recurrence. Expert Rev. Vaccines 2012, 11, 1315–1317. [Google Scholar] [CrossRef]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2018. [Google Scholar] [CrossRef]
- Hammerl, D.; Rieder, D.; Martens, J.W.M.; Trajanoski, Z.; Debets, R. Adoptive T Cell Therapy: New Avenues Leading to Safe Targets and Powerful Allies. Trends Immunol. 2018, 39, 921–936. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving cars to the clinic for solid tumors. Gene Ther. 2018. [Google Scholar] [CrossRef]
- Simon, B.; Uslu, U. CAR-T cell therapy in melanoma: A future success story? Exp. Dermatol. 2018, 27, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Hofflin, S.; Huckelhoven, A.; Bergmann, S.; Harrer, E.; Schuler, G.; Dorrie, J.; Schaft, N.; Harrer, T. Human T cells expressing two additional receptors (TETARs) specific for HIV-1 recognize both epitopes. Blood 2011, 118, 5174–5177. [Google Scholar] [CrossRef] [PubMed]
- Hofflin, S.; Prommersberger, S.; Uslu, U.; Schuler, G.; Schmidt, C.W.; Lennerz, V.; Dorrie, J.; Schaft, N. Generation of CD8(+) T cells expressing two additional T-cell receptors (TETARs) for personalised melanoma therapy. Cancer Biol. Ther. 2015, 16, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Hombach, A.; Sent, D.; Schneider, C.; Heuser, C.; Koch, D.; Pohl, C.; Seliger, B.; Abken, H. T-cell activation by recombinant receptors: CD28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 2001, 61, 1976–1982. [Google Scholar] [PubMed]
- Uslu, U.; Schuler, G.; Dorrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Dermatol. 2016, 25, 872–879. [Google Scholar] [CrossRef]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef]
- Riet, T.; Holzinger, A.; Dorrie, J.; Schaft, N.; Schuler, G.; Abken, H. Nonviral RNA transfection to transiently modify T cells with chimeric antigen receptors for adoptive therapy. Methods Mol. Biol. 2013, 969, 187–201. [Google Scholar] [PubMed]
- Bedoya, F.; Frigault, M.J.; Maus, M.V. The Flipside of the Power of Engineered T Cells: Observed and Potential Toxicities of Genetically Modified T Cells as Therapy. Mol. Ther. 2017, 25, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef]
- Chekenya, M.; Rooprai, H.K.; Davies, D.; Levine, J.M.; Butt, A.M.; Pilkington, G.J. The NG2 chondroitin sulfate proteoglycan: Role in malignant progression of human brain tumours. Int. J. Dev. Neurosci. 1999, 17, 421–435. [Google Scholar] [CrossRef]
- Godal, A.; Bruland, O.; Haug, E.; Aas, M.; Fodstad, O. Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br. J. Cancer 1986, 53, 839–841. [Google Scholar] [CrossRef]
- Ferrone, S.; Chen, Z.J.; Liu, C.C.; Hirai, S.; Kageshita, T.; Mittelman, A. Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol. Ther. 1993, 57, 259–290. [Google Scholar] [CrossRef]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Ferrone, S.; Ruiter, D.J. Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am. J. Pathol. 1990, 136, 1393–1405. [Google Scholar] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Spiotto, M.T.; Yu, P.; Rowley, D.A.; Nishimura, M.I.; Meredith, S.C.; Gajewski, T.F.; Fu, Y.X.; Schreiber, H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity 2002, 17, 737–747. [Google Scholar] [CrossRef]
- June, C.H.; Maus, M.V.; Plesa, G.; Johnson, L.A.; Zhao, Y.; Levine, B.L.; Grupp, S.A.; Porter, D.L. Engineered T cells for cancer therapy. Cancer Immunol. Immunother. 2014, 63, 969–975. [Google Scholar] [CrossRef]
- Boyiadzis, M.M.; Dhodapkar, M.V.; Brentjens, R.J.; Kochenderfer, J.N.; Neelapu, S.S.; Maus, M.V.; Porter, D.L.; Maloney, D.G.; Grupp, S.A.; Mackall, C.L.; et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: Clinical perspective and significance. J. Immunother. Cancer 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Slaney, C.Y.; von Scheidt, B.; Davenport, A.J.; Beavis, P.A.; Westwood, J.A.; Mardiana, S.; Tscharke, D.C.; Ellis, S.; Prince, H.M.; Trapani, J.A.; et al. Dual-specific Chimeric Antigen Receptor T Cells and an Indirect Vaccine Eradicate a Variety of Large Solid Tumors in an Immunocompetent, Self-antigen Setting. Clin. Cancer Res. 2017, 23, 2478–2490. [Google Scholar] [CrossRef]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef]
- Taussig, D.C.; Pearce, D.J.; Simpson, C.; Rohatiner, A.Z.; Lister, T.A.; Kelly, G.; Luongo, J.L.; Danet-Desnoyers, G.A.; Bonnet, D. Hematopoietic stem cells express multiple myeloid markers: Implications for the origin and targeted therapy of acute myeloid leukemia. Blood 2005, 106, 4086–4092. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schaft, N.; Schuler, G.; Dorrie, J.; Uslu, U. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in-vitro CAR-T cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp. Dermatol. 2018, 27, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dorrie, J.; Thumann, P.; Beck, V.E.; Muller, I.; Schultz, E.S.; Kampgen, E.; Dieckmann, D.; Schuler, G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J. Immunol. 2005, 174, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Wiesinger, M.; Marz, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schuler, G.; Uslu, U. Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers 2019, 11, 696. https://doi.org/10.3390/cancers11050696
Simon B, Harrer DC, Schuler-Thurner B, Schuler G, Uslu U. Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers. 2019; 11(5):696. https://doi.org/10.3390/cancers11050696
Chicago/Turabian StyleSimon, Bianca, Dennis C. Harrer, Beatrice Schuler-Thurner, Gerold Schuler, and Ugur Uslu. 2019. "Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer" Cancers 11, no. 5: 696. https://doi.org/10.3390/cancers11050696
APA StyleSimon, B., Harrer, D. C., Schuler-Thurner, B., Schuler, G., & Uslu, U. (2019). Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers, 11(5), 696. https://doi.org/10.3390/cancers11050696