The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sources of Intra-Tumoral Heterogeneity in Ovarian Cancer
2.1. Genetic Changes
2.2. Epigenetic Changes
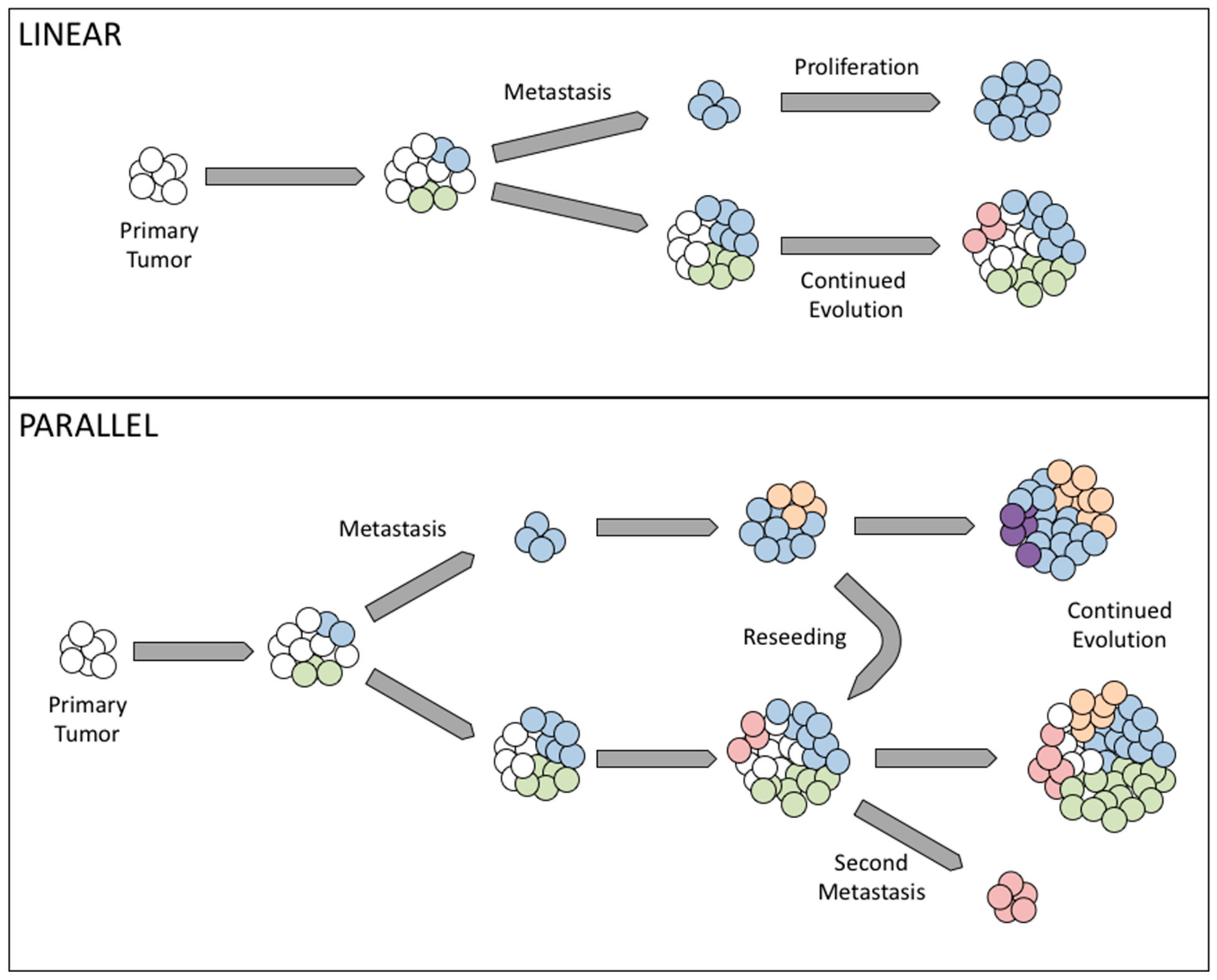
3. Clonal Evolution Theories in Ovarian Cancer
4. The Role of Cancer Stem Cells in Ovarian Cancer
4.1. Identifying Ovarian Cancer Stem Cells
4.2. Role of Cancer Stem Cells in Metastasis and Recurrence in Ovarian Cancer
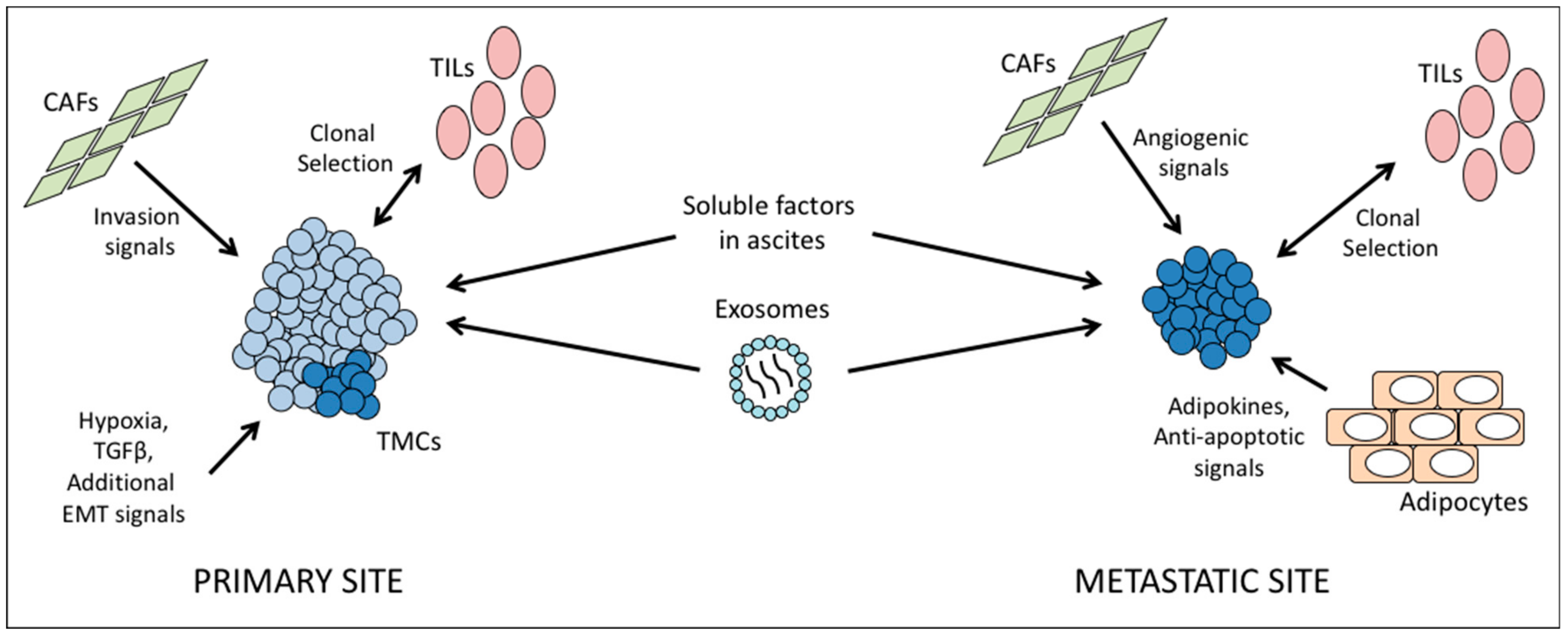
5. Biological Interactions within the Tumor Microenvironment
6. Clinical Relevance of Tumor Heterogeneity
7. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Can. J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Can. J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. Tumor Heterogeneity and the Biology of Cancer Invasion and Metastasis. Can. Res. 1978, 38, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Hallou, A.; Jennings, J.; Kabla Alexandre, J. Tumour heterogeneity promotes collective invasion and cancer metastatic dissemination. R. Soc. Open Sci. 2017, 4, 161007. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Shamai, Y.; Berger, L.; Skorecki, K.; Tzukerman, M. Niche-dependent gene expression profile of intratumoral heterogeneous ovarian cancer stem cell populations. PLoS ONE 2013, 8, e83651. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef]
- Bayani, J.; Paderova, J.; Murphy, J.; Rosen, B.; Zielenska, M.; Squire, J.A. Distinct patterns of structural and numerical chromosomal instability characterize sporadic ovarian cancer. Neoplasia 2008, 10, 1057–1065. [Google Scholar] [CrossRef]
- Penner-Goeke, S.; Lichtensztejn, Z.; Neufeld, M.; Ali, J.L.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. The temporal dynamics of chromosome instability in ovarian cancer cell lines and primary patient samples. PLoS Genet. 2017, 13, e1006707. [Google Scholar] [CrossRef]
- Swanton, C.; Nicke, B.; Schuett, M.; Eklund, A.C.; Ng, C.; Li, Q.; Hardcastle, T.; Lee, A.; Roy, R.; East, P.; et al. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 2009, 106, 8671–8676. [Google Scholar] [CrossRef]
- Singer, G.; Kallinowski, T.; Hartmann, A.; Dietmaier, W.; Wild, P.J.; Schraml, P.; Sauter, G.; Mihatsch, M.J.; Moch, H. Different types of microsatellite instability in ovarian carcinoma. Int. J. Cancer 2004, 112, 643–646. [Google Scholar] [CrossRef]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003, 34, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ru, N.; Xiao, H.; Chaturbedi, A.; Hoa, N.T.; Tian, X.J.; Zhang, H.; Ke, C.; Yan, F.; Nelson, J.; et al. Tumor-specific chromosome mis-segregation controls cancer plasticity by maintaining tumor heterogeneity. PLoS ONE 2013, 8, e80898. [Google Scholar] [CrossRef] [PubMed]
- Trinh, A.L.; Chen, H.; Chen, Y.; Hu, Y.; Li, Z.; Siegel, E.R.; Linskey, M.E.; Wang, P.H.; Digman, M.A.; Zhou, Y.H. Tracking Functional Tumor Cell Subpopulations of Malignant Glioma by Phasor Fluorescence Lifetime Imaging Microscopy of NADH. Cancers (Basel) 2017, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Natanzon, Y.; Goode, E.L.; Cunningham, J.M. Epigenetics in ovarian cancer. Semin Cancer Biol. 2018, 51, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Hyler, A.R.; Baudoin, N.C.; Brown, M.S.; Stremler, M.A.; Cimini, D.; Davalos, R.V.; Schmelz, E.M. Fluid shear stress impacts ovarian cancer cell viability, subcellular organization, and promotes genomic instability. PLoS ONE 2018, 13, e0194170. [Google Scholar] [CrossRef] [PubMed]
- Pejovic, T.; Yates, J.E.; Liu, H.Y.; Hays, L.E.; Akkari, Y.; Torimaru, Y.; Keeble, W.; Rathbun, R.K.; Rodgers, W.H.; Bale, A.E.; et al. Cytogenetic instability in ovarian epithelial cells from women at risk of ovarian cancer. Cancer Res. 2006, 66, 9017–9025. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Nat. Can. Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Breast Cancer Susceptibility, C.; Eccles, D.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Laufer, M.; Nandula, S.V.; Modi, A.P.; Wang, S.; Jasin, M.; Murty, V.V.V.S.; Ludwig, T.; Baer, R. Structural Requirements for the BARD1 Tumor Suppressor in Chromosomal Stability and Homology-directed DNA Repair. J. Biol. Chem. 2007, 282, 34325–34333. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, F.E.; Manley, J.L. The BARD1-CstF-50 Interaction Links mRNA 3’ End Formation to DNA Damage and Tumor Suppression. Cell 2001, 104, 743–753. [Google Scholar] [CrossRef]
- Metcalfe, K.A.; Akbari, M.R.; Narod, S.; Lerner-Ellis, J. PALB2 mutations in high-risk women with breast or ovarian cancer. J. Clin. Oncol. 2017, 35, 1527. [Google Scholar] [CrossRef]
- Pensabene, M.; Condello, C.; Carlomagno, C.; De Placido, S.; Liccardo, R.; Duraturo, F. Two novel sequence variants in MSH2 gene in a patient who underwent cancer genetic counseling for a very early-onset epithelial ovarian cancer. Hered. Can. Clin. Pract. 2016, 14, 18. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Decker, P.A.; Doffek, K.; Wang, B.; Xu, L.; Demeure, M.J.; Jen, J.; Sidransky, D. Microsatellite instability at selected tetranucleotide repeats is associated with p53 mutations in non-small cell lung cancer. Cancer Res. 2000, 60, 2488–2491. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; Van der Zee, A.G.J.; De Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yang, Y.; Zhou, N.; Tang, K.; Lau, W.B.; Lau, B.; Wang, W.; Xu, L.; Yang, Z.; Huang, S.; et al. Epigenetics in ovarian cancer: Premise, properties, and perspectives. Mol. Cancer 2018, 17, 109. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Haaf, T. Methylation Dynamics in the Early Mammalian Embryo: Implications of Genome Reprogramming Defects for Development. In DNA Methylation: Development, Genetic Disease and Cancer; Doerfler, W., Böhm, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 13–22. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG Island Methylation of DNA Damage Response Genes in Advanced Ovarian Cancer. Cancer Res. 2005, 65, 8961. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, M.; Mir, R.; Dholariya, S.; Najar, I.; Yadav, P.; Javid, J.; Guru, S.; Mirza, M.; Gandhi, G.; Khurana, N.; et al. RASSF1 and PTEN Promoter Hypermethylation Influences the Outcome in Epithelial Ovarian Cancer. Clin. Ovarian Other Gynecol. Cancer 2014, 7, 33–39. [Google Scholar] [CrossRef]
- Schöndorf, T.; Ebert, M.P.; Hoffmann, J.; Becker, M.; Moser, N.; Pur, Ş.; Göhring, U.-J.; Weisshaar, M.-P. Hypermethylation of the PTEN gene in ovarian cancer cell lines. Cancer Lett. 2004, 207, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, A.; Pisarcik, D.; Roberts, D.; Weinstein, J.; Cairns, P.; Hamilton, T.C. LOT1 (PLAGL1/ZAC1), the Candidate Tumor Suppressor Gene at Chromosome 6q24–25, Is Epigenetically Regulated in Cancer. J. Biol. Chem. 2003, 278, 6041–6049. [Google Scholar] [CrossRef]
- Häfner, N.; Diebolder, H.; Jansen, L.; Hoppe, I.; Dürst, M.; Runnebaum, I.B. Hypermethylated DAPK in serum DNA of women with uterine leiomyoma is a biomarker not restricted to cancer. Gynecol. Oncol. 2011, 121, 224–229. [Google Scholar] [CrossRef]
- Terasawa, K.; Sagae, S.; Toyota, M.; Tsukada, K.; Ogi, K.; Satoh, A.; Mita, H.; Imai, K.; Tokino, T.; Kudo, R. Epigenetic Inactivation of TMS1/ASC in Ovarian Cancer. Clin. Cancer Res. 2004, 10, 2000–2006. [Google Scholar] [CrossRef]
- Arnold, J.M.; Cummings, M.; Purdie, D.; Chenevix-Trench, G. Reduced expression of intercellular adhesion molecule-1 in ovarian adenocarcinomas. Br. J. Cancer 2001, 85, 1351–1358. [Google Scholar] [CrossRef]
- Yuecheng, Y.; Hongmei, L.; Xiaoyan, X. Clinical Evaluation of E-cadherin Expression and its Regulation Mechanism in Epithelial Ovarian Cancer. Clin. Exp. Metastasis 2006, 23, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Gayther, S.A.; Apostolidou, S.; Jones, A.; Lechner, M.; Beck, S.; Jacobs, I.J.; et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE 2009, 4, e8274. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jiang, G.; Woods, C.; Müller, H.M.; Fiegl, H.; Goebel, G.; Marth, C.; Müller-Holzner, E.; Zeimet, A.G.; Laird, P.W.; et al. DNA Hypomethylation and Ovarian Cancer Biology. Cancer Res. 2004, 64, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Cai, J.; Ding, H.; Xu, L.; Yang, Q.; Wang, Z. EZH2 participates in malignant biological behavior of epithelial ovarian cancer through regulating the expression of BRCA1. Cancer Biol. 2014, 15, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Hersey, J.M.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G.; et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer 2011, 10, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.Y.; Kim, K.S.; Hwang, S.H.; Kwon, K.S.; Kim, K.R.; Park, H.S.; Park, B.-H.; Chung, M.J.; Kang, M.J.; Lee, D.G.; et al. Expression and prognostic significance of SIRT1 in ovarian epithelial tumours. Pathology 2009, 41, 366–371. [Google Scholar] [CrossRef]
- Mvunta, D.H.; Miyamoto, T.; Asaka, R.; Yamada, Y.; Ando, H.; Higuchi, S.; Ida, K.; Kashima, H.; Shiozawa, T. Overexpression of SIRT1 is Associated With Poor Outcomes in Patients With Ovarian Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 415–421. [Google Scholar] [CrossRef]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Muller, P.; et al. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar] [CrossRef]
- Khalique, L.; Ayhan, A.; Whittaker, J.C.; Singh, N.; Jacobs, I.J.; Gayther, S.A.; Ramus, S.J. The clonal evolution of metastases from primary serous epithelial ovarian cancers. Int. J. Cancer 2009, 124, 1579–1586. [Google Scholar] [CrossRef]
- Klotz, D.M.; Wimberger, P. Cells of origin of ovarian cancer: Ovarian surface epithelium or fallopian tube? Arch. Gynecol. Obs. 2017, 296, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.; Iniesta, M.D.; Patrono, M.G.; Lu, K.H.; Ramirez, P.T. Role of Fallopian Tubes in the Development of Ovarian Cancer. J. Minim. Invasive Gynecol. 2017, 24, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massague, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.Q.; Kandyba, E.; Harris, S.; Del Rosario, R.; Balmain, A. Multicolour lineage tracing reveals clonal dynamics of squamous carcinoma evolution from initiation to metastasis. Nat. Cell Biol. 2018, 20, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Huntly, B.J.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 5, 311–321. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Badve, S.; Nakshatri, H. Breast-cancer stem cells-beyond semantics. Lancet Oncol. 2012, 13, 43–48. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Kalirai, H.; Clarke, R.B. Human breast epithelial stem cells and their regulation. J. Pathol. 2006, 208, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and Progenitor-Like Cells Contribute to the Aggressive Behavior of Human Epithelial Ovarian Cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2008, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 Expression Defines a Tumor Initiating Cell Population in Primary Human Ovarian Cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wu, P.Y. CD133 as a Marker for Cancer Stem Cells: Progresses and Concerns. Stem Cells Dev. 2009, 18, 1127–1134. [Google Scholar] [CrossRef]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde Dehydrogenase in Combination with CD133 Defines Angiogenic Ovarian Cancer Stem Cells That Portend Poor Patient Survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef]
- Alvero, A.B.; Chen, R.; Fu, H.-H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.-A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef]
- Alvero, A.B.; Fu, H.-H.; Holmberg, J.; Visintin, I.; Mor, L.; Marquina, C.C.; Oidtman, J.; Silasi, D.-A.; Mor, G. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 2009, 27, 2405–2413. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.M.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.Q.; Choi, Y.P.; Kang, S.; Youn, J.H.; Cho, N.H. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 2010, 29, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Parte, S.C.; Batra, S.K.; Kakar, S.S. Characterization of stem cell and cancer stem cell populations in ovary and ovarian tumors. J. Ovarian Res. 2018, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Terai, Y.; Tanabe, A.; Ono, Y.J.; Hayashi, M.; Maeda, K.; Fujiwara, S.; Ashihara, K.; Nakamura, M.; Tanaka, Y.; et al. CD24 expression is a marker for predicting clinical outcome and regulates the epithelial-mesenchymal transition in ovarian cancer via both the Akt and ERK pathways. Oncol. Rep. 2017, 37, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.; Chang, S.F.; Hsiao, C.C.; Chien, T.Y.; Shih, D.T. Isolation and characterization of stromal progenitor cells from ascites of patients with epithelial ovarian adenocarcinoma. J. Biomed. Sci. 2012, 19, 23. [Google Scholar] [CrossRef]
- Kenda Suster, N.; Smrkolj, S.; Virant-Klun, I. Putative stem cells and epithelial-mesenchymal transition revealed in sections of ovarian tumor in patients with serous ovarian carcinoma using immunohistochemistry for vimentin and pluripotency-related markers. J. Ovarian Res. 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Skorecki, K.; Tzukerman, M. Niche-dependent tumorigenic capacity of malignant ovarian ascites-derived cancer cell subpopulations. Clin. Cancer Res. 2009, 15, 70–80. [Google Scholar] [CrossRef]
- Yin, G.; Alvero, A.B.; Craveiro, V.; Holmberg, J.C.; Fu, H.H.; Montagna, M.K.; Yang, Y.; Chefetz-Menaker, I.; Nuti, S.; Rossi, M.; et al. Constitutive proteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene 2013, 32, 39–49. [Google Scholar] [CrossRef]
- Ovarian Tumor Tissue Analysis (OTTA) Consortium; Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; et al. Dose-Response Relationship of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef]
- Stanske, M.; Wienert, S.; Castillo-Tong, D.C.; Kreuzinger, C.; Vergote, I.; Lambrechts, S.; Gabra, H.; Gourley, C.; Ganapathi, R.N.; Kolaschinski, I.; et al. Dynamics of the Intratumoral Immune Response during Progression of High-Grade Serous Ovarian Cancer. Neoplasia 2018, 20, 280–288. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; De Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769. [Google Scholar] [CrossRef] [PubMed]
- Lis, R.; Touboul, C.; Halabi, N.M.; Madduri, A.S.; Querleu, D.; Mezey, J.; Malek, J.A.; Suhre, K.; Rafii, A. Mesenchymal cell interaction with ovarian cancer cells induces a background dependent pro-metastatic transcriptomic profile. J. Transl. Med. 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.; Cai, J.; Zhang, T.; Guo, J.; Feng, D.; Wang, Z. Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 2011, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Corvigno, S.; Wisman, G.B.; Mezheyeuski, A.; Van der Zee, A.G.; Nijman, H.W.; Avall-Lundqvist, E.; Ostman, A.; Dahlstrand, H. Markers of fibroblast-rich tumor stroma and perivascular cells in serous ovarian cancer: Inter- and intra-patient heterogeneity and impact on survival. Oncotarget 2016, 7, 18573–18584. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Montagna, M.K.; Pitruzzello, M.; Lima, E.; Mor, G.; Alvero, A.B. Adipocyte microenvironment promotes Bclxl expression and confers chemoresistance in ovarian cancer cells. Apoptosis 2017, 22, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Bhusari, P.A.; Khairnar, K.B. Greater Omental Pancake Tumour due to Metastasis of Ovarian Cancer—A Cadaveric Study. J. Clin. Diagn. Res. JCDR 2014, 8, 142–143. [Google Scholar] [CrossRef]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer–mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Nowicka, A.; Marini, F.C.; Solley, T.N.; Elizondo, P.B.; Zhang, Y.; Sharp, H.J.; Broaddus, R.; Kolonin, M.; Mok, S.C.; Thompson, M.S.; et al. Human omental-derived adipose stem cells increase ovarian cancer proliferation, migration, and chemoresistance. PLoS ONE 2013, 8, e81859. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Afrasiabi, K.; Linskey, M.E. Extracellular control of chromosomal instability and maintenance of intra-tumoral heterogeneity. J. Cancer Metastasis Treat. 2018, 4, 41. [Google Scholar] [CrossRef]
- Poste, G.; Doll, J.; Fidler, I.J. Interactions among clonal subpopulations affect stability of the metastatic phenotype in polyclonal populations of B16 melanoma cells. Proc. Natl. Acad. Sci. USA 1981, 78, 6226–6230. [Google Scholar] [CrossRef] [PubMed]
- Tedja, R.; Roberts, C.M.; Alvero, A.B.; Cardenas, C.; Yang-Hartwich, Y.; Spadinger, S.; Pitruzzello, M.; Yin, G.; Glackin, C.A.; Mor, G. Protein kinase Calpha-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J. Biol. Chem. 2019, 294, 5082–5093. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, B.; Song, Y.S. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016, 107, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Guo, N. Exosomes: Potent regulators of tumor malignancy and potential bio-tools in clinical application. Crit. Rev. Oncol. Hematol. 2015, 95, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Mor, G.; Cheng, H.; Xiang, X.; Hui, P.; Rutherford, T.; Yin, G.; Rimm, D.L.; Holmberg, J.; Alvero, A.; et al. High frequency of putative ovarian cancer stem cells with CD44/CK19 coexpression is associated with decreased progression-free intervals in patients with recurrent epithelial ovarian cancer. Reprod. Sci. 2013, 20, 605–615. [Google Scholar] [CrossRef]
- Steffensen, K.D.; Alvero, A.B.; Yang, Y.; Waldstrom, M.; Hui, P.; Holmberg, J.C.; Silasi, D.A.; Jakobsen, A.; Rutherford, T.; Mor, G. Prevalence of epithelial ovarian cancer stem cells correlates with recurrence in early-stage ovarian cancer. J. Oncol. 2011, 2011, 620523. [Google Scholar] [CrossRef]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-Molecule Ferroptotic Agents with Potential to Selectively Target Cancer Stem Cells. Sci. Rep. 2019, 9, 5926. [Google Scholar] [CrossRef]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle delivery of siRNA against TWIST to reduce drug resistance and tumor growth in ovarian cancer models. Nanomedicine 2016, 13, 965–976. [Google Scholar] [CrossRef]
- Shahin, S.A.; Wang, R.; Simargi, S.I.; Contreras, A.; Parra Echavarria, L.; Qu, L.; Wen, W.; Dellinger, T.; Unternaehrer, J.; Tamanoi, F.; et al. Hyaluronic acid conjugated nanoparticle delivery of siRNA against TWIST reduces tumor burden and enhances sensitivity to cisplatin in ovarian cancer. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Agliano, A.; Calvo, A.; Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin Cancer Biol. 2017, 44, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Gogola, E.; Rottenberg, S.; Jonkers, J. Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annu. Rev. Cancer Biol. 2019, 3, 235–254. [Google Scholar] [CrossRef]
- Leung, A.K. Poly(ADP-ribose): An organizer of cellular architecture. J. Cell Biol. 2014, 205, 613–619. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; Van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.E.; MacAuley, M.J.; Yadav, G.; Vizeacoumar, F.S.; Freywald, A.; Vizeacoumar, F.J. Targeting the CINful genome: Strategies to overcome tumor heterogeneity. Prog. Biophys. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cope, L.; Wu, R.C.; Shih Ie, M.; Wang, T.L. High level of chromosomal aberration in ovarian cancer genome correlates with poor clinical outcome. Gynecol. Oncol. 2013, 128, 500–505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Appierto, V.; Di Cosimo, S.; Reduzzi, C.; Pala, V.; Cappelletti, V.; Daidone, M.G. How to study and overcome tumor heterogeneity with circulating biomarkers: The breast cancer case. Semin Cancer Biol. 2017, 44, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Blassl, C.; Kuhlmann, J.D.; Webers, A.; Wimberger, P.; Fehm, T.; Neubauer, H. Gene expression profiling of single circulating tumor cells in ovarian cancer - Establishment of a multi-marker gene panel. Mol. Oncol. 2016, 10, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, R.; McCormick, A.; Mukhopadhyay, A.; Woodhouse, L.C.; Moat, M.; Grundy, A.; Dixon, M.; Kaufman, A.; Soohoo, S.; Elattar, A.; et al. The use of ovarian cancer cells from patients undergoing surgery to generate primary cultures capable of undergoing functional analysis. PLoS ONE 2014, 9, e90604. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. https://doi.org/10.3390/cancers11081083
Roberts CM, Cardenas C, Tedja R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers. 2019; 11(8):1083. https://doi.org/10.3390/cancers11081083
Chicago/Turabian StyleRoberts, Cai M., Carlos Cardenas, and Roslyn Tedja. 2019. "The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis" Cancers 11, no. 8: 1083. https://doi.org/10.3390/cancers11081083
APA StyleRoberts, C. M., Cardenas, C., & Tedja, R. (2019). The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers, 11(8), 1083. https://doi.org/10.3390/cancers11081083