Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2


Abstract
1. Introduction
2. Results
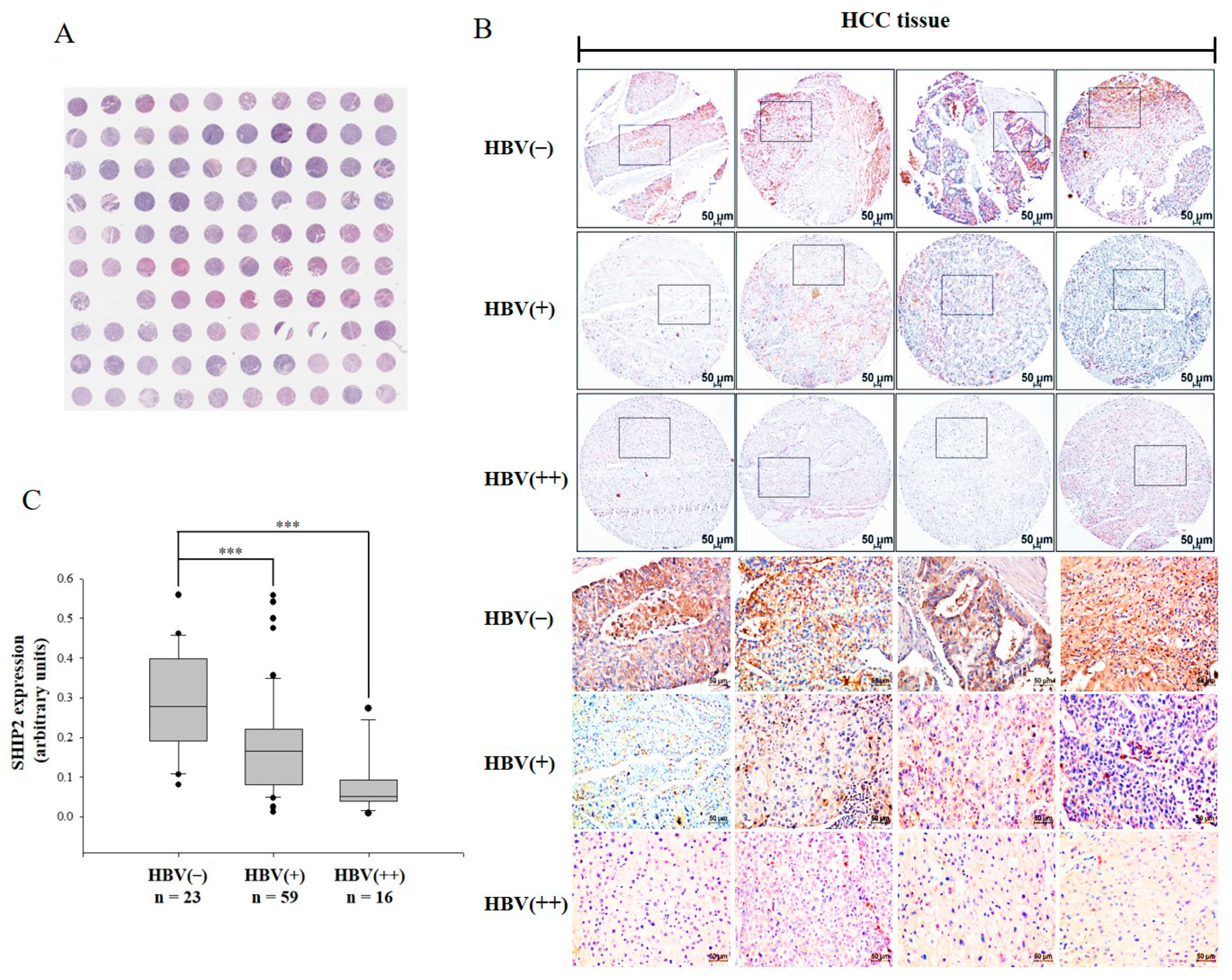
2.1. SHIP2 Is Downregulated in HBV-Infected HCC Tissues
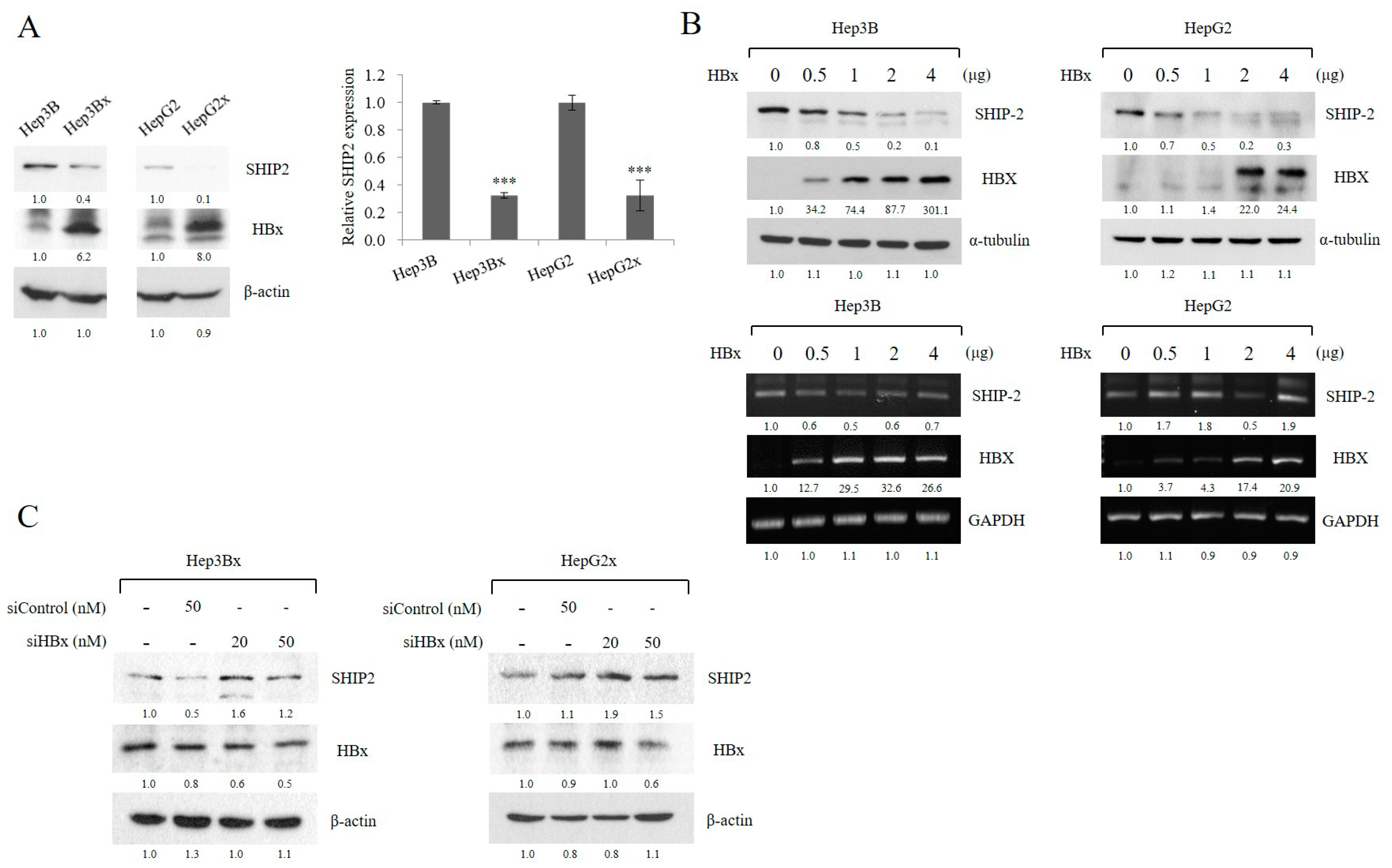
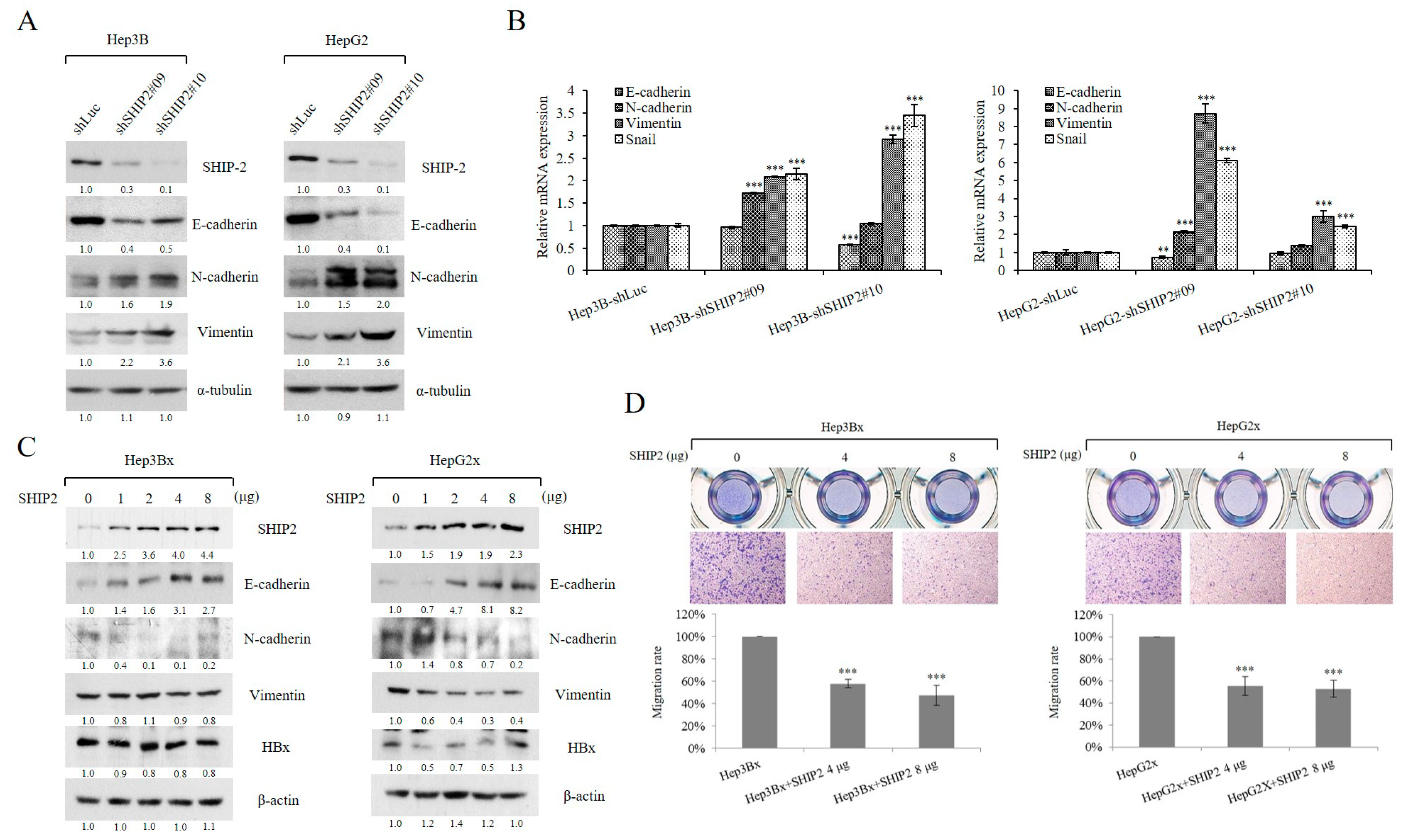
2.2. HBx Reduces SHIP2 Expression in HCC Cells
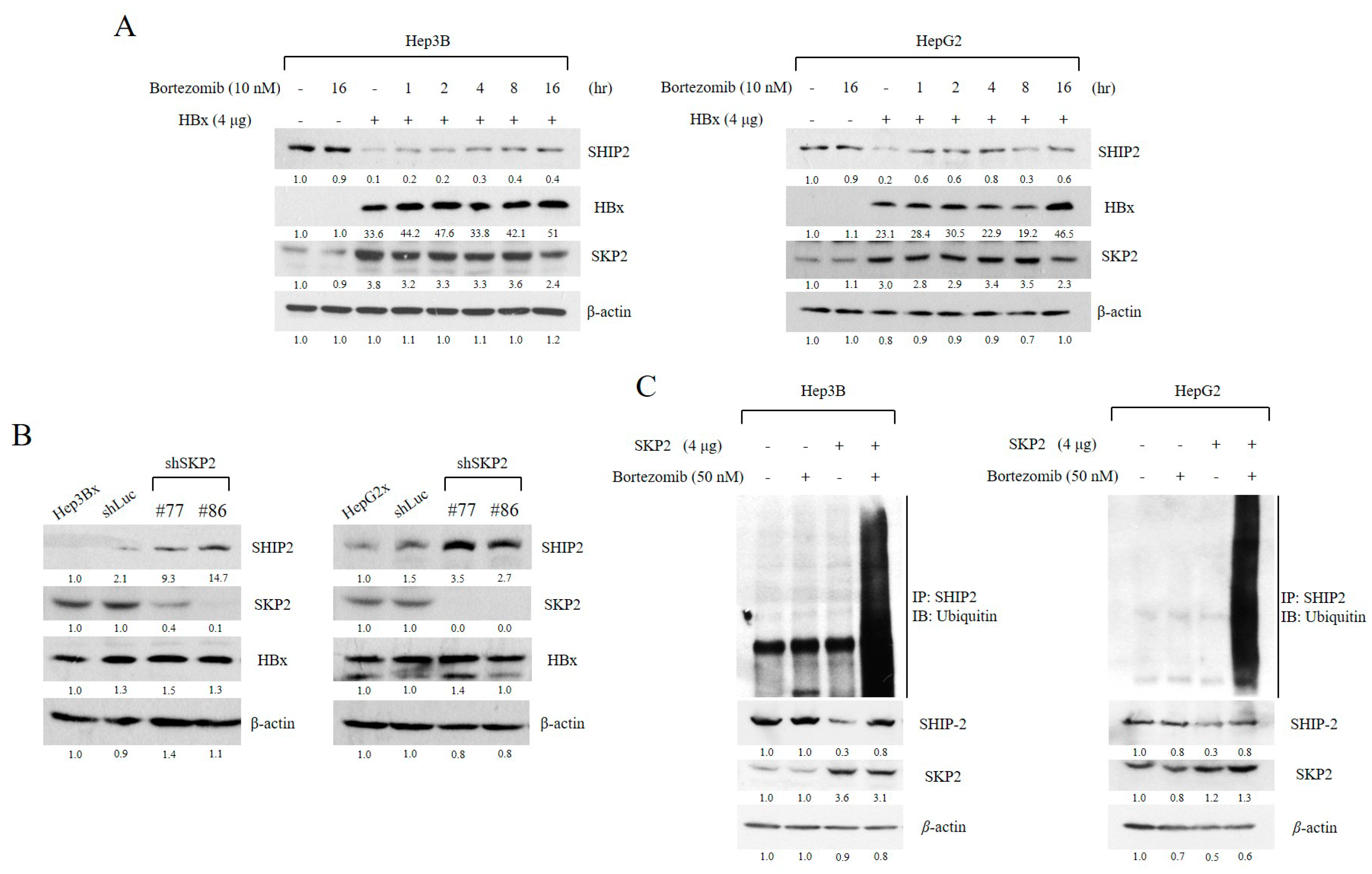
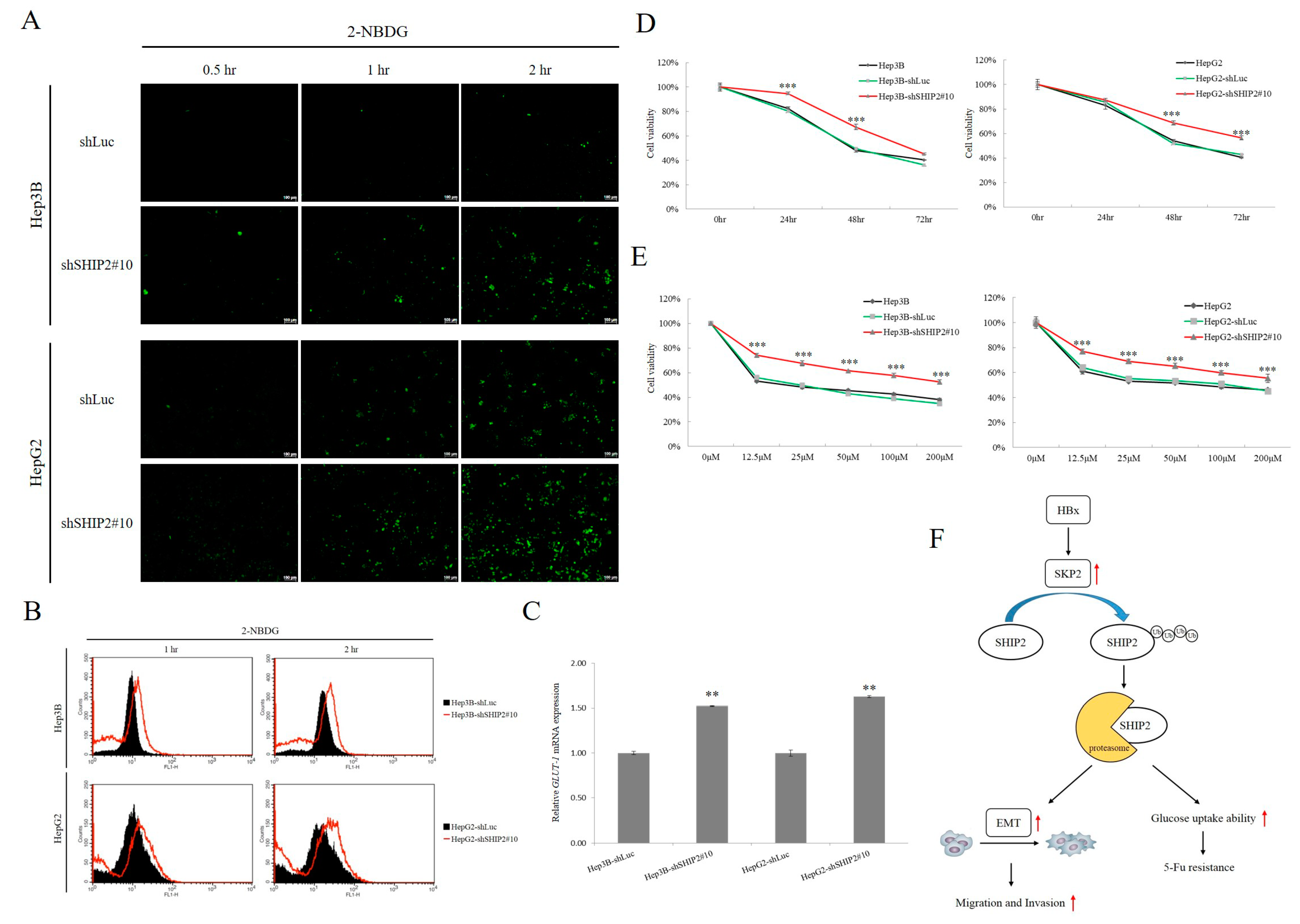
2.3. SHIP2 Is Regulated by HBx through SKP2 in HCC Cells
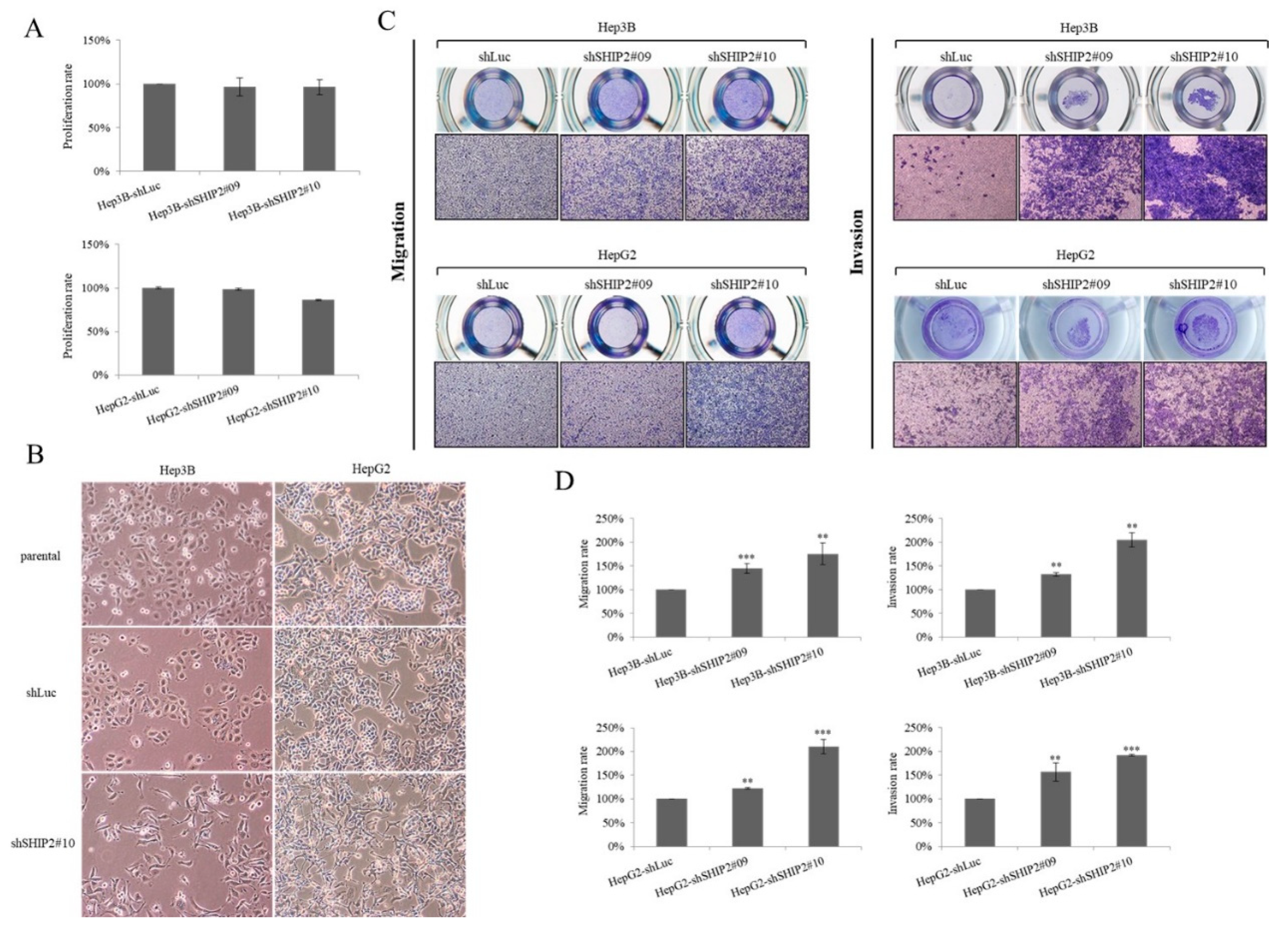
2.4. Downregulation of SHIP2 Leads to Elevated Glucose Uptake and Drug Resistance to 5-FU
3. Discussion
4. Materials and Methods
4.1. TMA Analysis
4.2. Cell Culture
4.3. Transient Transfection
4.4. Western Blot Analysis
4.5. RNA Isolation, Reverse Transcription, PCR, and Real-Time PCR
4.6. Immunoprecipitation
4.7. Gene Knockdown with shRNA
4.8. Cell Viability Assay
4.9. In Vitro Migration and Invasion Assays
4.10. Glucose Uptake Assay and Flow Cytometry Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kao, J.H.; Chen, D.S. Global control of hepatitis B virus infection. Lancet Infect. Dis. 2002, 2, 395–403. [Google Scholar] [CrossRef]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, S45–S55. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Schroder, C.H.; Hofmann, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology 1998, 27, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugan, M.; Moreno-Otero, R.; Sanz-Cameno, P. The molecular and pathophysiological implications of hepatitis B X antigen in chronic hepatitis B virus infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, L.; Wang, Q.W.; Yan, Q.X.; Yu, J.M. Effect of a conserved peptide derived from Kunitz domain of hepatitis B virus x protein on the cell cycle and apoptosis of HepG2 cells via the proteasome pathway. Chin. Med. J. 2009, 122, 460–465. [Google Scholar] [PubMed]
- Jung, J.K.; Kwun, H.J.; Lee, J.O.; Arora, P.; Jang, K.L. Hepatitis B virus X protein differentially affects the ubiquitin-mediated proteasomal degradation of beta-catenin depending on the status of cellular p53. J. Gen. Virol. 2007, 88, 2144–2154. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Chu, P.C.; Chuang, H.C.; Hung, W.C.; Kulp, S.K.; Chen, C.S. Targeting the oncogenic E3 ligase Skp2 in prostate and breast cancer cells with a novel energy restriction-mimetic agent. PLoS ONE 2012, 7, e47298. [Google Scholar] [CrossRef]
- Chen, F.Z.; Zhao, X.K. Ubiquitin-proteasome pathway and prostate cancer. Onkologie 2013, 36, 592–596. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, D.; Fukushima, H.; Inuzuka, H.; Liu, P.; Wan, L.; Sarkar, F.H.; Wei, W. Skp2: A novel potential therapeutic target for prostate cancer. Biochim. Biophys. Acta 2012, 1825, 11–17. [Google Scholar] [CrossRef]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef]
- Huang, Y.; Tong, S.; Tai, A.W.; Hussain, M.; Lok, A.S. Hepatitis B virus core promoter mutations contribute to hepatocarcinogenesis by deregulating SKP2 and its target, p21. Gastroenterology 2011, 141, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Lee, S.W.; Wang, J.; Lin, H.K. Regulation of Skp2 expression and activity and its role in cancer progression. Sci. World J. 2010, 10, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.C.; Pagano, M. Role of the F-box protein Skp2 in adhesion-dependent cell cycle progression. J. Cell Biol. 2001, 153, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.K.; Tandon, M.; Badve, S.; Snyder, P.W.; Nakshatri, H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis 2008, 29, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pesesse, X.; Deleu, S.; De Smedt, F.; Drayer, L.; Erneux, C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 1997, 239, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Krause, U.; Desmedt, F.; Tanti, J.F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef]
- Elong Edimo, W.; Derua, R.; Janssens, V.; Nakamura, T.; Vanderwinden, J.M.; Waelkens, E.; Erneux, C. Evidence of SHIP2 Ser132 phosphorylation, its nuclear localization and stability. Biochem. J. 2011, 439, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Erneux, C.; Edimo, W.E.; Deneubourg, L.; Pirson, I. SHIP2 multiple functions: A balance between a negative control of PtdIns(3,4,5)P(3) level, a positive control of PtdIns(3,4)P(2) production, and intrinsic docking properties. J. Cell. Biochem. 2011, 112, 2203–2209. [Google Scholar] [CrossRef]
- De Schutter, J.; Guillabert, A.; Imbault, V.; Degraef, C.; Erneux, C.; Communi, D.; Pirson, I. SHIP2 (SH2 domain-containing inositol phosphatase 2) SH2 domain negatively controls SHIP2 monoubiquitination in response to epidermal growth factor. J. Biol. Chem. 2009, 284, 36062–36076. [Google Scholar] [CrossRef]
- Grempler, R.; Zibrova, D.; Schoelch, C.; Van Marle, A.; Rippmann, J.F.; Redemann, N. Normalization of prandial blood glucose and improvement of glucose tolerance by liver-specific inhibition of SH2 domain containing inositol phosphatase 2 (SHIP2) in diabetic KKAy mice: SHIP2 inhibition causes insulin-mimetic effects on glycogen metabolism, gluconeogenesis, and glycolysis. Diabetes 2007, 56, 2235–2241. [Google Scholar] [CrossRef][Green Version]
- Akram, M. Mini-review on glycolysis and cancer. J. Cancer Educ. 2013, 28, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Sumie, S.; Kawaguchi, T.; Komuta, M.; Kuromatsu, R.; Itano, S.; Okuda, K.; Taniguchi, E.; Ando, E.; Takata, A.; Fukushima, N.; et al. Significance of glucose intolerance and SHIP2 expression in hepatocellular carcinoma patients with HCV infection. Oncol. Rep. 2007, 18, 545–552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taylor, V.; Wong, M.; Brandts, C.; Reilly, L.; Dean, N.M.; Cowsert, L.M.; Moodie, S.; Stokoe, D. 5′ phospholipid phosphatase SHIP-2 causes protein kinase B inactivation and cell cycle arrest in glioblastoma cells. Mol. Cell. Biol. 2000, 20, 6860–6871. [Google Scholar] [CrossRef] [PubMed]
- Kalra, N.; Kumar, V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett. 2006, 580, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. CMLS 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, Y.; Shen, Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J. Biochem. Biophys. Methods 2005, 64, 207–215. [Google Scholar] [CrossRef]
- Fuhler, G.M.; Brooks, R.; Toms, B.; Iyer, S.; Gengo, E.A.; Park, M.Y.; Gumbleton, M.; Viernes, D.R.; Chisholm, J.D.; Kerr, W.G. Therapeutic potential of SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol. Med. 2012, 18, 65–75. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Foti, M. PTEN and SHIP2 phosphoinositide phosphatases as negative regulators of insulin signalling. Arch. Physiol. Biochem. 2006, 112, 89–104. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Jurynec, M.J.; Grunwald, D.J. SHIP2, a factor associated with diet-induced obesity and insulin sensitivity, attenuates FGF signaling in vivo. Dis. Models Mech. 2010, 3, 733–742. [Google Scholar] [CrossRef]
- Elong Edimo, W.; Schurmans, S.; Roger, P.P.; Erneux, C. SHIP2 signaling in normal and pathological situations: Its impact on cell proliferation. Adv. Biol. Regul. 2014, 54, 142–151. [Google Scholar] [CrossRef]
- Yu, J.; Peng, H.; Ruan, Q.; Fatima, A.; Getsios, S.; Lavker, R.M. MicroRNA-205 promotes keratinocyte migration via the lipid phosphatase SHIP2. FASEB J. 2010, 24, 3950–3959. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.K. SHIP2 phosphoinositol phosphatase positively regulates EGFR-Akt pathway, CXCR4 expression, and cell migration in MDA-MB-231 breast cancer cells. Int. J. Oncol. 2009, 34, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lian, Z.; Han, S.; Waye, M.M.; Wang, H.; Wu, M.C.; Wu, K.; Ding, J.; Arbuthnot, P.; Kew, M.; et al. Downregulation of E-cadherin by hepatitis B virus X antigen in hepatocellullar carcinoma. Oncogene 2006, 25, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.; Sar, S.; Barre, R.; Cariven, C.; Marin, M.; Salles, J.P.; Erneux, C.; Samuel, D.; Gassama-Diagne, A. SHIP2 regulates epithelial cell polarity through its lipid product, which binds to Dlg1, a pathway subverted by hepatitis C virus core protein. Mol. Biol. Cell 2013, 24, 2171–2185. [Google Scholar] [CrossRef]
- Motavaf, M.; Safari, S.; Saffari Jourshari, M.; Alavian, S.M. Hepatitis B virus-induced hepatocellular carcinoma: The role of the virus x protein. Acta Virol. 2013, 57, 389–396. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Yan, J.; Yao, Z.; Hu, K.; Zhong, Y.; Li, M.; Xiong, Z.; Deng, M. Hepatitis B Virus Core Promoter A1762T/G1764A (TA)/T1753A/T1768A Mutations Contribute to Hepatocarcinogenesis by Deregulating Skp2 and P53. Dig. Dis. Sci. 2015, 60, 1315–1324. [Google Scholar] [CrossRef]
- Suwa, A.; Kurama, T.; Yamamoto, T.; Sawada, A.; Shimokawa, T.; Aramori, I. Glucose metabolism activation by SHIP2 inhibitors via up-regulation of GLUT1 gene in L6 myotubes. Eur. J. Pharmacol. 2010, 642, 177–182. [Google Scholar] [CrossRef]
- Wada, T.; Sasaoka, T.; Funaki, M.; Hori, H.; Murakami, S.; Ishiki, M.; Haruta, T.; Asano, T.; Ogawa, W.; Ishihara, H.; et al. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol. Cell. Biol. 2001, 21, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-D-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Marinari, B.; Moretti, F.; Botti, E.; Giustizieri, M.L.; Descargues, P.; Giunta, A.; Stolfi, C.; Ballaro, C.; Papoutsaki, M.; Alema, S.; et al. The tumor suppressor activity of IKKalpha in stratified epithelia is exerted in part via the TGF-beta antiproliferative pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 17091–17096. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKbeta to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef] [PubMed]
- Hsia, T.C.; Tu, C.Y.; Chen, Y.J.; Wei, Y.L.; Yu, M.C.; Hsu, S.C.; Tsai, S.L.; Chen, W.S.; Yeh, M.H.; Yen, C.J.; et al. Lapatinib-mediated cyclooxygenase-2 expression via epidermal growth factor receptor/HuR interaction enhances the aggressiveness of triple-negative breast cancer cells. Mol. Pharmacol. 2013, 83, 857–869. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | No. (%) |
|---|---|
| Gender, n (%) | |
| Male | 42 (85.7) |
| Female | 7 (14.3) |
| Age (years) | |
| Median (range) | 50 (24–68) |
| Stage, n (%) | |
| I | 5 (10.2) |
| II | 42 (85.7) |
| III | 2 (4) |
| HBV infection, n (%) | |
| − | 23 (23.5) |
| + | 59 (60.2) |
| ++ | 16 (16.3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, K.-J.; Yu, Y.-L. Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2. Cancers 2019, 11, 1065. https://doi.org/10.3390/cancers11081065
Su K-J, Yu Y-L. Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2. Cancers. 2019; 11(8):1065. https://doi.org/10.3390/cancers11081065
Chicago/Turabian StyleSu, Kuo-Jung, and Yung-Luen Yu. 2019. "Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2" Cancers 11, no. 8: 1065. https://doi.org/10.3390/cancers11081065
APA StyleSu, K.-J., & Yu, Y.-L. (2019). Downregulation of SHIP2 by Hepatitis B Virus X Promotes the Metastasis and Chemoresistance of Hepatocellular Carcinoma through SKP2. Cancers, 11(8), 1065. https://doi.org/10.3390/cancers11081065