The Unsolved Puzzle of c-Rel in B Cell Lymphoma

Abstract
1. Introduction: c-Rel Is the NF-κB Family Transcription Factor with the Strongest Link to Human Lymphoma
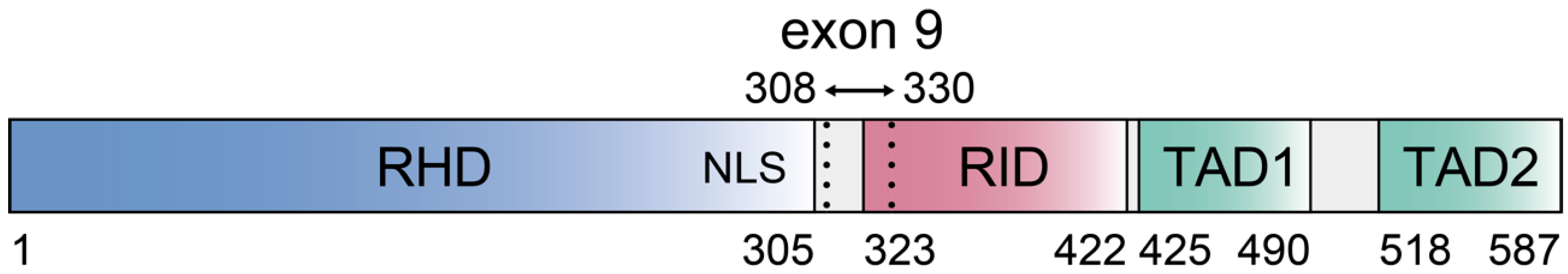
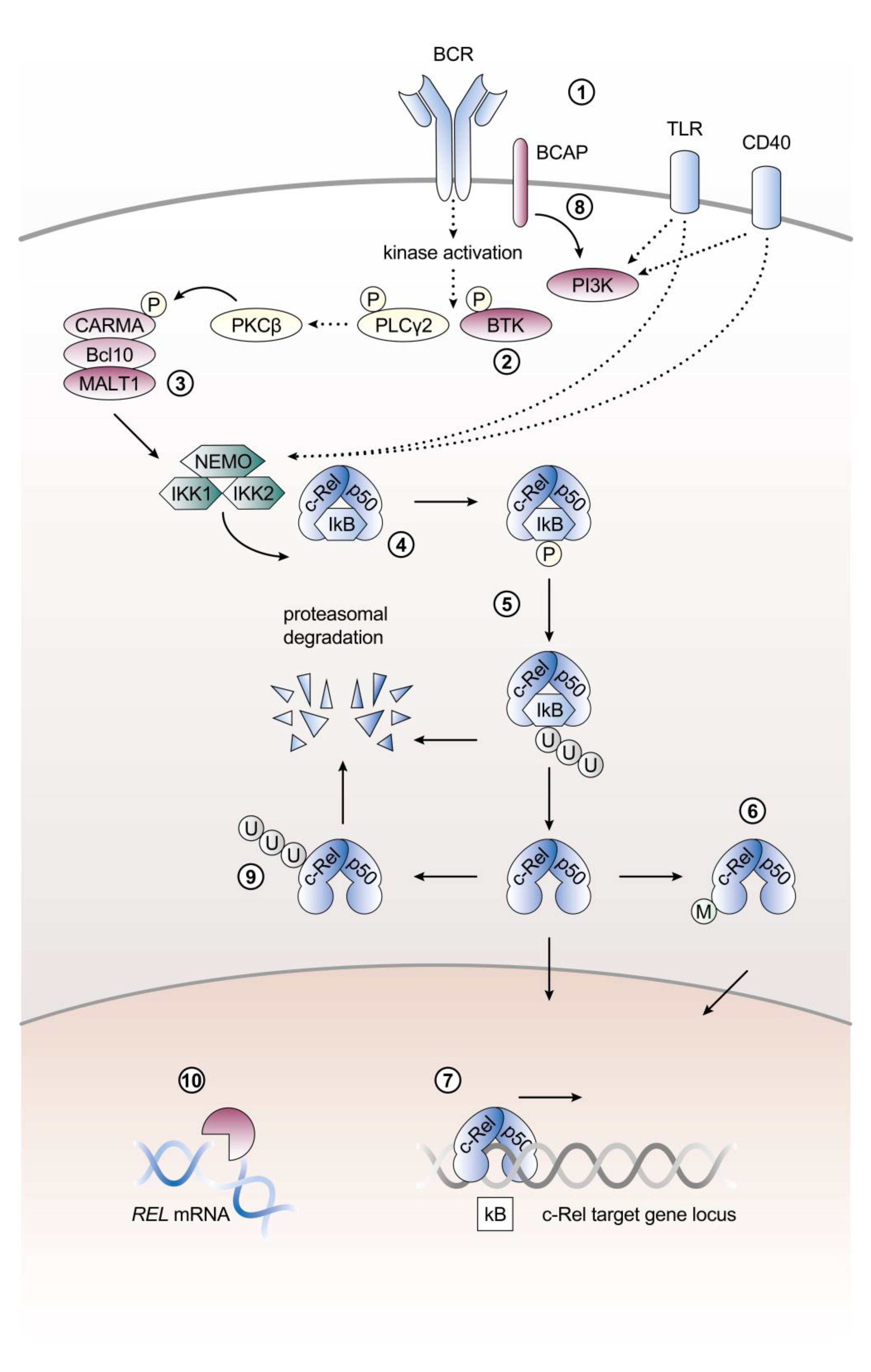
2. Control of c-Rel Expression, Abundance, and Activation in B Cells
3. Aspects of B Cell Lymphomagenesis
4. Genetic Aberrations Involving the REL Gene Locus in B Cell Lymphoma
4.1. REL Gains in Diffuse Large B Cell Lymphoma
4.2. REL Gains in Follicular Lymphoma and Transformed Follicular Lymphoma
4.3. REL Gains in Primary Mediastinal B Cell Lymphoma
4.4. REL Gains in Classical Hodgkin Lymphoma
5. REL Gain in Relation to c-Rel Protein Abundance and Localization
5.1. Expression and Localization of c-Rel in DLBCL
5.2. Localization of c-Rel in PMBCL
5.3. Localization of c-Rel in cHL
6. BCL11A Is Frequently Co-Amplified with REL
7. Investigating the Oncogenic Role of c-Rel in Mouse Models
8. c-Rel as a Therapeutic Target
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Smale, S.T. Selectivity of the NF-κB Response. Cold Spring Harb. Perspect. Biol. 2010, 2, a000257. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.; Weih, F.; Bravo, R. Developmental expression of the mouse c-rel proto-oncogene in hematopoietic organs. Development 1994, 120, 2991–3004. [Google Scholar] [PubMed]
- Kontgen, F.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Li, R.; Tarlinton, D.; Gerondakis, S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995, 9, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Tumang, J.R.; Owyang, A.; Andjelic, S.; Jin, Z.; Hardy, R.R.; Liou, M.L.; Liou, H.C. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur. J. Immunol. 1998, 28, 4299–4312. [Google Scholar] [CrossRef]
- Harling-McNabb, L.; Deliyannis, G.; Jackson, D.C.; Gerondakis, S.; Grigoriadis, G.; Brown, L.E. Mice lacking the transcription factor subunit Rel can clear an influenza infection and have functional anti-viral cytotoxic T cells but do not develop an optimal antibody response. Int. Immunol. 1999, 11, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Heise, N.; De Silva, N.S.; Silva, K.; Carette, A.; Simonetti, G.; Pasparakis, M.; Klein, U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J. Exp. Med. 2014, 211, 2103–2118. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-κB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hayden, M.S. New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef]
- Staudt, L.M. Oncogenic Activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef]
- Lim, K.-H.; Yang, Y.; Staudt, L.M. Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunol. Rev. 2012, 246, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Cormier, C.; Jean-Jacques, J.; Gapuzan, M.E. Malignant transformation of primary chicken spleen cells by human transcription factor c-Rel. Oncogene 2001, 20, 7098–7103. [Google Scholar] [CrossRef] [PubMed]
- Brownell, E.; O’Brien, S.J.; Nash, W.G.; Rice, N. Genetic characterization of human c-rel sequences. Mol. Cell. Biol. 1985, 5, 2826–2831. [Google Scholar] [CrossRef] [PubMed]
- Leeman, J.R.; Weniger, M.A.; Barth, T.F.; Gilmore, T.D. Deletion analysis and alternative splicing define a transactivation inhibitory domain in human oncoprotein REL. Oncogene 2008, 27, 6770–6781. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, A.G.; San-Antonio, B.; Fresno, M. Regulation of nuclear factor κB transactivation. Implication of phosphatidylinositol 3-kinase and protein kinase C ζ in c-Rel activation by tumor necrosis factor α. J. Biol. Chem. 2001, 276, 15840–15849. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Reynolds, J.G.; Gilmore, T.D. Deletion of either C-terminal transactivation subdomain enhances the in vitro transforming activity of human transcription factor REL in chicken spleen cells. Oncogene 2003, 22, 6928–6936. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tam, W.F.; Wang, W.; Sen, R. Cell-specific association and shuttling of IκBα provides a mechanism for nuclear NF-κB in B lymphocytes. Mol. Cell. Biol. 2001, 21, 4837–4846. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, R.; Melén, K.; Cao, X.; Julkunen, I. NF-κB p52, RelB and c-Rel are transported into the nucleus via a subset of importin α molecules. Cell. Signal. 2008, 20, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Leeman, J.R.; Gilmore, T.D. Alternative splicing in the NF-κB signaling pathway. Gene 2008, 423, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Yu, M.; Mendoza, L.; Yunis, J.J. Cloning and transcription factor-binding sites of the human c-rel proto-oncogene promoter. Gene 1996, 170, 271–276. [Google Scholar] [CrossRef]
- Hu, C.J.; Rao, S.; Ramirez-Bergeron, D.L.; Garrett-Sinha, L.A.; Gerondakis, S.; Clark, M.R.; Simon, M.C. PU.1/Spi-B regulation of c-rel is essential for mature B cell survival. Immunity 2001, 15, 545–555. [Google Scholar] [CrossRef]
- Grumont, R.J.; Richardson, I.B.; Gaff, C.; Gerondakis, S. rel/NF-κB nuclear complexes that bind kB sites in the murine c-rel promoter are required for constitutive c-rel transcription in B-cells. Cell Growth Differ. 1993, 4, 731–743. [Google Scholar]
- Kaltschmidt, B.; Greiner, J.F.W.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Wuerzberger-Davis, S.M.; Chen, Y.; Yang, D.T.; Kearns, J.D.; Bates, P.W.; Lynch, C.; Ladell, N.C.; Yu, M.; Podd, A.; Zeng, H.; et al. Nuclear export of the NF-κB inhibitor IκBα is required for proper B cell and secondary lymphoid tissue formation. Immunity 2011, 34, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Terauchi, Y.; Fujiwara, M.; Aizawa, S.; Yazaki, Y.; Kadowaki, T.; Koyasu, S. Xid-Like Immunodeficiency in Mice with Disruption of the p85α Subunit of Phosphoinositide 3-Kinase. Science 1999, 283, 390–392. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kurosaki, T. Contribution of BCAP to maintenance of mature B cells through c-Rel. Nat. Immunol. 2003, 4, 780–786. [Google Scholar] [CrossRef]
- Matsuda, S.; Mikami, Y.; Ohtani, M.; Fujiwara, M.; Hirata, Y.; Minowa, A.; Terauchi, Y.; Kadowaki, T.; Koyasu, S. Critical role of class IA PI3K for c-Rel expression in B lymphocytes. Blood 2008, 113, 1037–1044. [Google Scholar] [CrossRef]
- Ferch, U.; Büschenfelde, C.M.Z.; Gewies, A.; Wegener, E.; Rauser, S.; Peschel, C.; Krappmann, D.; Ruland, J. MALT1 directs B cell receptor–induced canonical nuclear factor-κB signaling selectively to the c-Rel subunit. Nat. Immunol. 2007, 8, 984–991. [Google Scholar] [CrossRef]
- Shi, W.; Liao, Y.; Willis, S.N.; Taubenheim, N.; Inouye, M.; Tarlinton, D.M.; Smyth, G.K.; Hodgkin, P.D.; Nutt, S.L.; Corcoran, L.M. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat. Immunol. 2015, 16, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Tarte, K.; Zhan, F.; De Vos, J.; Klein, B.; Shaughnessy, J. Gene expression profiling of plasma cells and plasmablasts: Toward a better understanding of the late stages of B-cell differentiation. Blood 2003, 102, 592–600. [Google Scholar] [CrossRef] [PubMed]
- De Silva, N.S.; Anderson, M.M.; Carette, A.; Silva, K.; Heise, N.; Bhagat, G.; Klein, U. Transcription factors of the alternative NF-κB pathway are required for germinal center B-cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Heng, T.S.P.; Painter, M.W. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Mitchell, S.; Liu, Y.; Ohta, S.; Lin, Y.-S.; Metzig, M.O.; Nutt, S.L.; Hoffmann, A. A Regulatory Circuit Controlling the Dynamics of NFκB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity 2019, 50, 616–628.e6. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.E.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Möoller, P. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Klein, U.; Niu, H.; Stolovitzky, G.A.; Tu, Y.; Califano, A.; Cattoretti, G.; Dalla-Favera, R. Tracking CD40 signaling during germinal center development. Blood 2004, 104, 4088–4096. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-κB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Luedde, T.; Schmidt-Supprian, M. Dissection of the NF-κB signalling cascade in transgenic and knockout mice. Cell Death Differ. 2006, 13, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Kaileh, M.; Sen, R. NF-κB function in B lymphocytes. Immunol. Rev. 2012, 246, 254–271. [Google Scholar] [CrossRef]
- Jaworski, M.; Thome, M. The paracaspase MALT1: Biological function and potential for therapeutic inhibition. Cell. Mol. Life Sci. 2016, 73, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Fontan, L.; Yang, C.; Kabaleeswaran, V.; Volpon, L.; Osborne, M.J.; Beltran, E.; Garcia, M.; Cerchietti, L.; Shaknovich, R.; Yang, S.N.; et al. MALT1 Small Molecule Inhibitors Specifically Suppress ABC-DLBCL In Vitro and In Vivo. Cancer Cell 2012, 22, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Shinners, N.P.; Carlesso, G.; Castro, I.; Hoek, K.L.; Corn, R.A.; Woodland, R.T.; Woodland, R.L.; Scott, M.L.; Wang, D.; Khan, W.N. Bruton’s tyrosine kinase mediates NF-κB activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J. Immunol. 2007, 179, 3872–3880. [Google Scholar] [CrossRef] [PubMed]
- Damdinsuren, B.; Zhang, Y.; Khalil, A.; Wood, W.H.; Becker, K.G.; Shlomchik, M.J.; Sen, R. Single round of antigen receptor signaling programs naive B cells to receive T cell help. Immunity 2010, 32, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Liou, H.-C.; Hsia, C.Y. Distinctions between c-Rel and other NF-κB proteins in immunity and disease. Bioessays 2003, 25, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Aleksiyadis, K.; Martin, A.; McNamee, K.; Tharmalingam, T.; Williams, R.O.; Mémet, S.; Cope, A.P. Inhibitor of kappa B epsilon (IκBε) is a non-redundant regulator of c-Rel-dependent gene expression in murine T and B cells. PLoS ONE 2011, 6, e24504. [Google Scholar] [CrossRef] [PubMed]
- Alves, B.N.; Tsui, R.; Almaden, J.; Shokhirev, M.N.; Davis-Turak, J.; Fujimoto, J.; Birnbaum, H.; Ponomarenko, J.; Hoffmann, A. IκBε is a key regulator of B cell expansion by providing negative feedback on cRel and RelA in a stimulus-specific manner. J. Immunol. 2014, 192, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Uehata, T.; Iwasaki, H.; Vandenbon, A.; Matsushita, K.; Hernandez-Cuellar, E.; Kuniyoshi, K.; Satoh, T.; Mino, T.; Suzuki, Y.; Standley, D.M.; et al. Malt1-Induced Cleavage of Regnase-1 in CD4+ helper T cells regulates immune activation. Cell 2013, 153, 1036–1049. [Google Scholar] [CrossRef]
- Jeltsch, K.M.; Hu, D.; Brenner, S.; Zöller, J.; Heinz, G.A.; Nagel, D.; Vogel, K.U.; Rehage, N.; Warth, S.C.; Edelmann, S.L.; et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat. Immunol. 2014, 15, 1079–1089. [Google Scholar] [CrossRef]
- Bertossi, A.; Aichinger, M.; Sansonetti, P.; Lech, M.; Neff, F.; Pal, M.; Wunderlich, F.T.; Anders, H.-J.; Klein, L.; Schmidt-Supprian, M. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J. Exp. Med. 2011, 208, 1749–1756. [Google Scholar] [CrossRef]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Valdepeñas, C.; Martin, A.G.; Ramakrishnan, P.; Wallach, D.; Fresno, M. NF-κB-inducing kinase is involved in the activation of the CD28 responsive element through phosphorylation of c-Rel and regulation of its transactivating activity. J. Immunol. 2006, 176, 4666–4674. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Olière, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mitani, T.; Yorita, K.; Uchida, D.; Matsushima, A.; Iwamasa, K.; Fujita, S.; Matsumoto, M. Abnormal Immune Function of Hemopoietic Cells from Alymphoplasia (aly) Mice, a Natural Strain with Mutant NF-κB-Inducing Kinase. J. Immunol. 2000, 165, 804–812. [Google Scholar] [CrossRef]
- Martin, A.G.; Fresno, M. Tumor necrosis factor-α activation of NF-κB requires the phosphorylation of Ser-471 in the transactivation domain of c-Rel. J. Biol. Chem. 2000, 275, 24383–24391. [Google Scholar] [CrossRef]
- Fognani, C.; Rondi, R.; Romano, A.; Blasi, F. cRel-TD kinase: A serine/threonine kinase binding in vivo and in vitro c-Rel and phosphorylating its transactivation domain. Oncogene 2000, 19, 2224–2232. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Reynolds, J.G.; Gilmore, T.D. Mutations of tumor necrosis factor α-responsive serine residues within the C-terminal transactivation domain of human transcription factor REL enhance its in vitro transforming ability. Oncogene 2005, 24, 7355–7368. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Trautmann, H.; Pott, C.; Harder, L.; Arnold, N.; Africa, J.A.; Leeman, J.R.; Siebert, R.; Gilmore, T.D. Mutation of an IKK phosphorylation site within the transactivation domain of REL in two patients with B-cell lymphoma enhances REL’s in vitro transforming activity. Oncogene 2007, 26, 2685–2694. [Google Scholar] [CrossRef]
- Glineur, C.; Davioud-Charvet, E.; Vandenbunder, B. The conserved redox-sensitive cysteine residue of the DNA-binding region in the c-Rel protein is involved in the regulation of the phosphorylation of the protein. Biochem. J. 2000, 352, 583–591. [Google Scholar] [CrossRef]
- Gapuzan, M.E.R.; Pitoc, G.A.; Gilmore, T.D. Mutations within a conserved protein kinase A recognition sequence confer temperature-sensitive and partially defective activities onto mouse c-Rel. Biochem. Biophys. Res. Commun. 2003, 307, 92–99. [Google Scholar] [CrossRef]
- Chen, E.; Hrdlicková, R.; Nehyba, J.; Longo, D.L.; Bose, H.R.; Li, C.C. Degradation of proto-oncoprotein c-Rel by the ubiquitin-proteasome pathway. J. Biol. Chem. 1998, 273, 35201–35207. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Chang, J.-H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.-H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the Transcriptional Function of the NF-κB Protein c-Rel by O-GlcNAc Glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Fan, Y.; Gupta, N.; Matsuura, I.; Liu, F.; Zhou, X.Z.; Lu, K.P.; Gelinas, C. Peptidyl-Prolyl Isomerase Pin1 Markedly Enhances the Oncogenic Activity of the Rel Proteins in the Nuclear Factor-κB Family. Cancer Res. 2009, 69, 4589–4597. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Siebenlist, U.; Brown, K.; Claudio, E. Control of lymphocyte development by nuclear factor-κB. Nat. Rev. Immunol. 2005, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Travers, P.; Walport, M. Janeway’s Immunobiology, 7th ed.; Garland Publishing: New York, NY, USA, 2007. [Google Scholar]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Shen, H.M.; Peters, A.; Baron, B.; Zhu, X.; Storb, U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 1998, 280, 1750–1752. [Google Scholar] [CrossRef]
- Liu, M.; Duke, J.L.; Richter, D.J.; Vinuesa, C.G.; Goodnow, C.C.; Kleinstein, S.H.; Schatz, D.G. Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008, 451, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Rosenwald, A.; Staudt, L.M. Lymphoid malignancies: The dark side of B-cell differentiation. Nat. Rev. Immunol. 2002, 2, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Hömig-Hölzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune Surveillance and Therapy of Lymphomas Driven by Epstein-Barr Virus Protein LMP1 in a Mouse Model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rödling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, J.; Olshen, A.B.; Cattoretti, G.; Donnelly, G.B.; Teruya-Feldstein, J.; Qin, J.; Palanisamy, N.; Shen, Y.; Dyomina, K.; Petlakh, M.; et al. Relationship between REL amplification, REL function, and clinical and biologic features in diffuse large B-cell lymphomas. Blood 2004, 103, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Döring, C.; Vucic, E.; Chan, F.C.; Ennishi, D.; Tousseyn, T.; de Wolf-Peeters, C.; Perner, S.; Wlodarska, I.; Steidl, C.; et al. Array comparative genomic hybridization reveals similarities between nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma. Br. J. Haematol. 2015, 169, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, D.; Pantic, M.; Skatulla, I.; Rawluk, J.; Kreutz, C.; Martens, U.M.; Fisch, P.; Timmer, J.; Veelken, H. Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays. Blood 2007, 109, 1202–1210. [Google Scholar] [CrossRef]
- Hartmann, S.; Gesk, S.; Scholtysik, R.; Kreuz, M.; Bug, S.; Vater, I.; Döring, C.; Cogliatti, S.; Parrens, M.; Merlio, J.-P.; et al. High resolution SNP array genomic profiling of peripheral T cell lymphomas, not otherwise specified, identifies a subgroup with chromosomal aberrations affecting the REL locus. Br. J. Haematol. 2010, 148, 402–412. [Google Scholar] [CrossRef]
- Ramos, J.C.; Ruiz, P.; Ratner, L.; Reis, I.M.; Brites, C.; Pedroso, C.; Byrne, G.E.; Toomey, N.L.; Andela, V.; Harhaj, E.W.; et al. IRF-4 and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood 2007, 109, 3060–3068. [Google Scholar] [CrossRef]
- Reader, J.C.; Zhao, X.F.; Butler, M.S.; Rapoport, A.P.; Ning, Y. REL-positive double minute chromosomes in follicular lymphoma. Leukemia 2006, 20, 1624–1626. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Klapper, W.; Sotnikova, A.; Callet-Bauchu, E.; Harder, L.; Bastard, C.; Schmitz, R.; Grohmann, S.; Höppner, J.; Riemke, J.; et al. Deutsche Krebshilfe Network Project Molecular Mechanisms in Malignant Lymphomas Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res. 2006, 66, 10332–10338. [Google Scholar] [CrossRef] [PubMed]
- Enciso-Mora, V.; Broderick, P.; Ma, Y.; Jarrett, R.F.; Hjalgrim, H.; Hemminki, K.; van den Berg, A.; Olver, B.; Lloyd, A.; Dobbins, S.E.; et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat. Genet. 2010, 42, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Frampton, M.; da Silva Filho, M.I.; Broderick, P.; Thomsen, H.; Försti, A.; Vijayakrishnan, J.; Cooke, R.; Enciso-Mora, V.; Hoffmann, P.; Nöthen, M.M.; et al. Variation at 3p24.1 and 6q23.3 influences the risk of Hodgkin’s lymphoma. Nat. Commun. 2013, 4, 2549. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Gelinas, C. An optimal range of transcription potency is necessary for efficient cell transformation by c-Rel to ensure optimal nuclear localization and gene-specific activation. Oncogene 2007, 26, 4038–4043. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Houldsworth, J.; Mathew, S.; Rao, P.H.; Dyomina, K.; Louie, D.C.; Parsa, N.; Offit, K.; Chaganti, R.S. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood 1996, 87, 25–29. [Google Scholar]
- Rao, P.H.; Houldsworth, J.; Dyomina, K.; Parsa, N.Z.; Cigudosa, J.C.; Louie, D.C.; Popplewell, L.; Offit, K.; Jhanwar, S.C.; Chaganti, R.S. Chromosomal and gene amplification in diffuse large B-cell lymphoma. Blood 1998, 92, 234–240. [Google Scholar]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. Lymphoma/Leukemia Molecular Profiling Project The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Shipp, M.A.; Ross, K.N.; Tamayo, P.; Weng, A.P.; Kutok, J.L.; Aguiar, R.C.T.; Gaasenbeek, M.; Angelo, M.; Reich, M.; Pinkus, G.S.; et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med. 2002, 8, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.V.; Ewton, A.A.; Olsen, R.J.; Logan, B.R.; Preti, H.A.; Liu, Y.-C.; Perkins, S.L.; Chang, C.-C. Prognostic impact of C-REL expression in diffuse large B-cell lymphoma. J. Hematopathol. 2009, 2, 20–26. [Google Scholar] [CrossRef]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.T.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef]
- Bea, S.; Zettl, A.; Wright, G.; Salaverria, I.; Jehn, P.; Moreno, V.; Burek, C.; Ott, G.; Puig, X.; Yang, L.; et al. Lymphoma/Leukemia Molecular Profiling Project Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005, 106, 3183–3190. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.W.; Emre, N.C.T.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef]
- Feuerhake, F.; Kutok, J.L.; Monti, S.; Chen, W.; LaCasce, A.S.; Cattoretti, G.; Kurtin, P.; Pinkus, G.S.; de Leval, L.; Harris, N.L.; et al. NFκB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood 2005, 106, 1392–1399. [Google Scholar] [CrossRef]
- Tagawa, H.; Suguro, M.; Tsuzuki, S.; Matsuo, K.; Karnan, S.; Ohshima, K.; Okamoto, M.; Morishima, Y.; Nakamura, S.; Seto, M. Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood 2005, 106, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Colomo, L.; López-Guillermo, A.; Salaverria, I.; Puig, X.; Pinyol, M.; Rives, S.; Montserrat, E.; Campo, E. Clinicopathologic significance and prognostic value of chromosomal imbalances in diffuse large B-cell lymphomas. J. Clin. Oncol. 2004, 22, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Sehn, L.H.; Gascoyne, R.D. Pathogenesis of follicular lymphoma. J. Clin. Investig. 2012, 122, 3424–3431. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Sujobert, P.; Salles, G. From genetics to the clinic: A translational perspective on follicular lymphoma. Nat. Rev. Cancer 2018, 18, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hernandez, A.M.; Shibata, D.; Cortopassi, G.A. BCL2 translocation frequency rises with age in humans. Proc. Natl. Acad. Sci. USA 1994, 91, 8910–8914. [Google Scholar] [CrossRef] [PubMed]
- Nagy, M.; Balázs, M.; Adám, Z.; Petkó, Z.; Tímár, B.; Szereday, Z.; László, T.; Warnke, R.A.; Matolcsy, A. Genetic instability is associated with histological transformation of follicle center lymphoma. Leukemia 2000, 14, 2142–2148. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hough, R.E.; Goepel, J.R.; Alcock, H.E.; Hancock, B.W.; Lorigan, P.C.; Hammond, D.W. Copy number gain at 12q12-14 may be important in the transformation from follicular lymphoma to diffuse large B cell lymphoma. Br. J. Cancer 2001, 84, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.K.; Neat, M.J.; Crawley, C.R.; Jones, L.; Jones, E.; Lister, T.A.; Gupta, R.K. The use of real-time quantitative polymerase chain reaction and comparative genomic hybridization to identify amplification of the REL gene in follicular lymphoma. Br. J. Haematol. 2000, 111, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Climent, J.A.; Alizadeh, A.A.; Segraves, R.; Blesa, D.; Rubio-Moscardo, F.; Albertson, D.G.; Garcia-Conde, J.; Dyer, M.J.S.; Levy, R.; Pinkel, D.; et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood 2003, 101, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.J.; Rosenwald, A.; Wright, G.; Lee, A.; Last, K.W.; Weisenburger, D.D.; Chan, W.C.; Delabie, J.; Braziel, R.M.; Campo, E.; et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br. J. Haematol. 2007, 136, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbò, F.; et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinska, A.; Ichimura, K.; Berglund, M.; Dinets, A.; Sulaiman, L.; Collins, V.P.; Larsson, C.; Porwit, A.; Lagercrantz, S.B. Amplification of 2p as a genomic marker for transformation in lymphoma. Genes Chromosom. Cancer 2014, 53, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; McKeithan, T.W.; Deffenbacher, K.E.; Lachel, C.; Wright, G.W.; Iqbal, J.; Smith, L.M.; Zhang, W.; Kucuk, C.; Rinaldi, A.; et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood 2014, 123, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.; Leithäuser, F.; Joos, S.; Bentz, M.; Möller, P. Mediastinal (thymic) large B-cell lymphoma: Where do we stand? Lancet Oncol. 2002, 3, 229–234. [Google Scholar] [CrossRef]
- Savage, K.J. Primary mediastinal large B-cell lymphoma. Oncologist 2006, 11, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.S.; Otaño-Joos, M.I.M.; Ziegler, S.S.; Brüderlein, S.S.; du Manoir, S.S.; Bentz, M.M.; Möller, P.P.; Lichter, P.P. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 1996, 87, 1571–1578. [Google Scholar] [PubMed]
- Palanisamy, N.; Abou-Elella, A.A.; Chaganti, S.R.; Houldsworth, J.; Offit, K.; Louie, D.C.; Terayu-Feldstein, J.; Cigudosa, J.C.; Rao, P.H.; Sanger, W.G.; et al. Similar patterns of genomic alterations characterize primary mediastinal large-B-cell lymphoma and diffuse large-B-cell lymphoma. Genes Chromosom. Cancer 2002, 33, 114–122. [Google Scholar] [CrossRef]
- Bentz, M.; Barth, T.F.; Brüderlein, S.; Bock, D.; Schwerer, M.J.; Baudis, M.; Joos, S.; Viardot, A.; Feller, A.C.; Müller-Hermelink, H.K.; et al. Gain of chromosome arm 9p is characteristic of primary mediastinal B-cell lymphoma (MBL): Comprehensive molecular cytogenetic analysis and presentation of a novel MBL cell line. Genes Chromosom. Cancer 2001, 30, 393–401. [Google Scholar] [CrossRef]
- Weniger, M.A.; Gesk, S.; Ehrlich, S.; Martin-Subero, J.I.; Dyer, M.J.S.; Siebert, R.; Möller, P.; Barth, T.F.E. Gains of REL in primary mediastinal B-cell lymphoma coincide with nuclear accumulation of REL protein. Genes Chromosom. Cancer 2007, 46, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. New insights in the biology of Hodgkin lymphoma. ASH Educ. Program Book 2012, 2012, 328–334. [Google Scholar]
- Joos, S.; Menz, C.K.; Wrobel, G.; Siebert, R.; Gesk, S.; Ohl, S.; Mechtersheimer, G.; Trümper, L.; Möller, P.; Lichter, P.; et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002, 99, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Telenius, A.; Shah, S.P.; Farinha, P.; Barclay, L.; Boyle, M.; Connors, J.M.; Horsman, D.E.; Gascoyne, R.D. Genome-wide copy number analysis of Hodgkin Reed-Sternberg cells identifies recurrent imbalances with correlations to treatment outcome. Blood 2010, 116, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Adey, A.; Thomas, A.; Lee, C.; Liu, Y.J.; Kumar, A.; Lewis, A.P.; Wu, D.; Fromm, J.R.; Shendure, J. Recurrent somatic loss of TNFRSF14 in classical Hodgkin lymphoma. Genes Chromosom. Cancer 2016, 55, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F.; Jais, J.-P.; Molina, T.-J.; Parmentier, F.; Picquenot, J.-M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.-A.; Feugier, P.; et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: A GELA study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Odqvist, L.; Montes-Moreno, S.; Sánchez-Pacheco, R.E.; Young, K.H.; Martín-Sánchez, E.; Cereceda, L.; Sánchez-Verde, L.; Pajares, R.; Mollejo, M.; Fresno, M.F.; et al. NFκB expression is a feature of both activated B-cell-like and germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. Mod. Pathol. 2014, 27, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu-Monette, Z.Y.; Ok, C.Y.; Tzankov, A.; Manyam, G.C.; Sun, R.; Visco, C.; Zhang, M.; Montes-Moreno, S.; Dybkaer, K.; et al. Prognostic impact of c-Rel nuclear expression and REL amplification and crosstalk between c-Rel and the p53 pathway in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 23157–23180. [Google Scholar] [CrossRef]
- Rodig, S.J.; Savage, K.J.; LaCasce, A.S.; Weng, A.P.; Harris, N.L.; Shipp, M.A.; Hsi, E.D.; Gascoyne, R.D.; Kutok, J.L. Expression of TRAF1 and nuclear c-Rel distinguishes primary mediastinal large cell lymphoma from other types of diffuse large B-cell lymphoma. Am. J. Surg. Pathol. 2007, 31, 106–112. [Google Scholar] [CrossRef]
- Pham, L.V.; Fu, L.; Tamayo, A.T.; Bueso-Ramos, C.; Drakos, E.; Vega, F.; Medeiros, L.J.; Ford, R.J. Constitutive BR3 receptor signaling in diffuse, large B-cell lymphomas stabilizes nuclear factor-κB-inducing kinase while activating both canonical and alternative nuclear factor-κB pathways. Blood 2011, 117, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Savage, K.J.; Nguyen, V.; Pinkus, G.S.; Shipp, M.A.; Aster, J.C.; Kutok, J.L. TRAF1 expression and c-Rel activation are useful adjuncts in distinguishing classical Hodgkin lymphoma from a subset of morphologically or immunophenotypically similar lymphomas. Am. J. Surg. Pathol. 2005, 29, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Shen, N.; Hedvat, C.V.; Moskowitz, C.H.; Sussman, L.K.; Filippa, D.A.; Zelenetz, A.D.; Houldsworth, J.; Chaganti, R.S.K.; Teruya-Feldstein, J. Differential expression patterns of c-REL protein in classic and nodular lymphocyte predominant Hodgkin lymphoma. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 211–215. [Google Scholar]
- Gilmore, T.D.; Starczynowski, D.T.; Kalaitzidis, D. RELevant gene amplification in B-cell lymphomas? Blood 2004, 103, 3243–3245. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, E. The BCL11 gene family: Involvement of BCL11A in lymphoid malignancies. Blood 2001, 98, 3413–3420. [Google Scholar] [CrossRef]
- Fukuhara, N.; Tagawa, H.; Kameoka, Y.; Kasugai, Y.; Karnan, S.; Kameoka, J.; Sasaki, T.; Morishima, Y.; Nakamura, S.; Seto, M. Characterization of target genes at the 2p15-16 amplicon in diffuse large B-cell lymphoma. Cancer Sci. 2006, 97, 499–504. [Google Scholar] [CrossRef]
- Weniger, M.A.; Pulford, K.; Gesk, S.; Ehrlich, S.; Banham, A.H.; Lyne, L.; Martin-Subero, J.I.; Siebert, R.; Dyer, M.J.S.; Moller, P.; et al. Gains of the proto-oncogene BCL11A and nuclear accumulation of BCL11AXL protein are frequent in primary mediastinal B-cell lymphoma. Leukemia 2006, 20, 1880–1882. [Google Scholar] [CrossRef][Green Version]
- Weber, J.; de la Rosa, J.; Grove, C.S.; Schick, M.; Rad, L.; Baranov, O.; Strong, A.; Pfaus, A.; Friedrich, M.J.; Engleitner, T.; et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat. Commun. 2019, 10, 1415. [Google Scholar] [CrossRef]
- Hunter, J.E.; Butterworth, J.A.; Zhao, B.; Sellier, H.; Campbell, K.J.; Thomas, H.D.; Bacon, C.M.; Cockell, S.J.; Gewurz, B.E.; Perkins, N.D. The NF-κB subunit c-Rel regulates Bach2 tumour suppressor expression in B-cell lymphoma. Oncogene 2015, 35, 3476. [Google Scholar] [CrossRef]
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The Eμ-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med. 1988, 167, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Wang, S.; Yu, L.; Yi, H.; Liu, R.; Geng, W.; Wan, X.; Ma, Y.; Cai, L.; Chen, Y.H.; et al. Treating psoriasis by targeting its susceptibility gene Rel. Clin. Immunol. 2016, 165, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Regulation of tissue homeostasis by NF-κB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Garbati, M.R. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr. Top. Microbiol. Immunol. 2011, 349, 245–263. [Google Scholar] [PubMed]
- Ranuncolo, S.M.; Pittaluga, S.; Evbuomwan, M.O.; Jaffe, E.S.; Lewis, B.A. Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 2012, 120, 3756–3763. [Google Scholar] [CrossRef]
- Tian, W.; Liou, H.-C. RNAi-Mediated c-Rel Silencing Leads to Apoptosis of B Cell Tumor Cells and Suppresses Antigenic Immune Response In Vivo. PLoS ONE 2009, 4, e5028. [Google Scholar] [CrossRef]
- Yamamoto, M.; Horie, R.; Takeiri, M.; Kozawa, I.; Umezawa, K. Inactivation of NF-κB components by covalent binding of (-)-dehydroxymethylepoxyquinomicin to specific cysteine residues. J. Med. Chem. 2008, 51, 5780–5788. [Google Scholar] [CrossRef]
- Ouk, S.; Liou, M.-L.; Liou, H.-C. Direct Rel/NF-κB inhibitors: Structural basis for mechanism of action. Future Med. Chem. 2009, 1, 1683–1707. [Google Scholar] [CrossRef]
- Yeo, A.T.; Chennamadhavuni, S.; Whitty, A.; Porco, J.A.; Gilmore, T.D. Inhibition of Oncogenic Transcription Factor REL by the Natural Product Derivative Calafianin Monomer 101 Induces Proliferation Arrest and Apoptosis in Human B-Lymphoma Cell Lines. Molecules 2015, 20, 7474–7494. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Tuckett, A.Z.; Ouk, S.; Liou, H.-C.; Altan-Bonnet, G.; Tsai, J.J.; Oyler, J.E.; Smith, O.M.; West, M.L.; Singer, N.V.; et al. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising antitumor activity. Cancer Discov. 2014, 4, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Tuckett, A.Z.; Liou, H.-C.; Doubrovina, E.; Derenzini, E.; Ouk, S.; Tsai, J.J.; Smith, O.M.; Levy, E.R.; Kreines, F.M.; et al. Characterization of a c-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-κB-Controlled Oxidative Stress Responses. Cancer Res. 2016, 76, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Vaisitti, T.; Gaudino, F.; Ouk, S.; Moscvin, M.; Vitale, N.; Serra, S.; Arruga, F.; Zakrzewski, J.L.; Liou, H.-C.; Allan, J.N.; et al. Targeting metabolism and survival in chronic lymphocytic leukemia and Richter syndrome cells by a novel NF-κB inhibitor. Haematologica 2017, 102, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Amos, C.I.; Lee, A.T.; Lu, Y.; Remmers, E.F.; Kastner, D.L.; Seldin, M.F.; Criswell, L.A.; Plenge, R.M.; Holers, V.M.; et al. REL, encoding a member of the NF-κB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 2009, 41, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Eyre, S.; Hinks, A.; Flynn, E.; Martin, P.; Wilson, A.G.; Maxwell, J.R.; Morgan, A.W.; Emery, P.; Steer, S.; Hocking, L.J.; et al. Confirmation of association of the REL locus with rheumatoid arthritis susceptibility in the UK population. Ann. Rheum. Dis. 2010, 69, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Strange, A.; Capon, F.; Spencer, C.C.A.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.M.; et al. Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP. Nat. Genet. 2010, 42, 985–990. [Google Scholar]
- McGovern, D.P.B.; Gardet, A.; Törkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.H.; Lagacé, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef]
- Trynka, G.; Zhernakova, A.; Romanos, J.; Franke, L.; Hunt, K.A.; Turner, G.; Bruinenberg, M.; Heap, G.A.; Platteel, M.; Ryan, A.W.; et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-κB signalling. Gut 2009, 58, 1078–1083. [Google Scholar] [CrossRef]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Ádány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Sanz, I.; Cook, M.C. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009, 9, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The risk of lymphoma development in autoimmune diseases: A meta-analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Gadalla, S.; Morton, L.M.; Landgren, O.; Pfeiffer, R.; Warren, J.L.; Berndt, S.I.; Ricker, W.; Parsons, R.; Engels, E.A. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int. J. Cancer 2009, 125, 398–405. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Method | FL | tFL | DLBCL | GCB-DLBCL | ABC-DLBCL | PMBCL | cHL |
|---|---|---|---|---|---|---|---|---|
| Nagy et al. [107] | CGH | 0% (0/5) | 20% (1/5) | |||||
| Hough et al. [108] | CGH | 0% (0/18) | 17% (4/23) | 33% (6/18) | ||||
| Goff et al. [109] *1 | CGH, qPCR | 17% (1/6) | 50% (3/6) | |||||
| Martinez-Climent et al. [110] | aCGH | 10% (1/10) | 20% (2/10) | |||||
| Davies et al. [111] | qPCR | 5% (1/20) | 10% (2/20) | |||||
| Pasqualucci et al. [112] *2 | WES, SNP array | 33% (4/12) | 50% (6/12) 31% (12/39) | |||||
| Kwiecinska et al. [113] *3 | aCGH, qPCR | 20% (3/15) | 41% (12/29) | 3% (1/29) | ||||
| Bouska et al. [114] | SNP array | 24% (48/198) | 30% (24/79) | |||||
| Houldsworth et al. [89] Rao et al. [90] | SB | 23% (26/111) *4 | ||||||
| Bea et al. [103] | CGH | 14% (9/64) | ||||||
| Jardin et al. [128] | QMPSF | 22% (5/23) *5 | 37% (10/27) | 9% (6/64) | ||||
| Rosenwald et al. [94] *6 | qPCR | 15% (17/115) | 0% (0/73) | |||||
| Bea et al. [99] *6 | CGH | 17% (15/87) | 15% (12/77) | 47% (9/19) | ||||
| Lenz et al. [100] *6 | aCGH | 35% (72) | 12% (74) | 26% (31) | ||||
| Houldsworth et al. [77] | SB | 28% (5/18) | 17% (2/12) | |||||
| Feuerhake et al. [101] | qPCR | 17% (10/57) | 5% (1/22) | 3% (1/34) | ||||
| Tagawa et al. [102] | aCGH | 33% (6/18) | 7% (2/28) | |||||
| Joos et al. [119] | CGH | 27% (7/26) | ||||||
| Bentz et al. [121] *7 | CGH | 19% (8/43) | ||||||
| Weniger et al. [122] *7 | FISH | 75% (15/20) | ||||||
| Palanisamy et al. [120] | SB | 36% (4/11) | ||||||
| Martin-Subero et al. [83] | FICTION | 35% (11/31) | ||||||
| Joos et al. [125] *8 | CGH | 54% (22/41) | ||||||
| Barth et al. [36] *8 | CGH, FICTION | 41% (7/17) | ||||||
| Steidl et al. [126] | aCGH | 28% (53) | ||||||
| Salipante et al. [127] | WGS | 40% (8/20) | ||||||
| Average | 20% | 30% | 17% | 21% | 9% | 28% | 39% |
| Reference | Method | FL | tFL | DLBCL | GCB-DLBCL | ABC-DLBCL | PMBCL | cHL |
|---|---|---|---|---|---|---|---|---|
| Martinez-Climent et al. [110] | aCGH | 0% (0/10) | 20% (2/10) | |||||
| Pasqualucci et al. [112] | WES, SNP array | 8% (1/12) | 8% (1/12) 31% (12/39) | |||||
| Kwiecinska et al. [113] *1 | aCGH, qPCR | 7% (1/15) | 20% (3/15) | |||||
| Bouska et al. [114] | SNP array | 10% (19/198) | 13% (10/79) | |||||
| Houldsworth et al. [89] Rao et al. [90] | SB | 23% (26/111) | ||||||
| Jardin et al. [128] *2 | QMPSF | 4% (1/23) | 7% (2/27) | 2% (1/64) | ||||
| Rosenwald et al. [94] | qPCR | 15% (17/115) | 0% (0/73) | |||||
| Houldsworth et al. [77] | SB | 28% (5/18) | 17% (2/12) | |||||
| Feuerhake et al. [101] | qPCR | 17% (10/57) | 5% (1/22) | 3% (1/34) | ||||
| Joos et al. [119] | CGH | 8% (2/26) | ||||||
| Bentz et al. [121] | CGH | 0% (0/43) | ||||||
| Weniger et al. [122] | FISH | 25% (5/20) | ||||||
| Palanisamy et al. [120] | SB | 36% (4/11) | ||||||
| Martin-Subero et al. [83] | FICTION | 26% (8/31) | ||||||
| Joos et al. [125] | CGH | 2% (1/41) | ||||||
| Barth et al. [36] | CGH, FICTION | 12% (2/17) | ||||||
| Salipante et al. [127] | WGS | 30% (6/20) | ||||||
| Average | 9% | 18% | 12% | 16% | 2% | 9% | 16% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kober-Hasslacher, M.; Schmidt-Supprian, M. The Unsolved Puzzle of c-Rel in B Cell Lymphoma. Cancers 2019, 11, 941. https://doi.org/10.3390/cancers11070941
Kober-Hasslacher M, Schmidt-Supprian M. The Unsolved Puzzle of c-Rel in B Cell Lymphoma. Cancers. 2019; 11(7):941. https://doi.org/10.3390/cancers11070941
Chicago/Turabian StyleKober-Hasslacher, Maike, and Marc Schmidt-Supprian. 2019. "The Unsolved Puzzle of c-Rel in B Cell Lymphoma" Cancers 11, no. 7: 941. https://doi.org/10.3390/cancers11070941
APA StyleKober-Hasslacher, M., & Schmidt-Supprian, M. (2019). The Unsolved Puzzle of c-Rel in B Cell Lymphoma. Cancers, 11(7), 941. https://doi.org/10.3390/cancers11070941