NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB


Abstract
1. Introduction
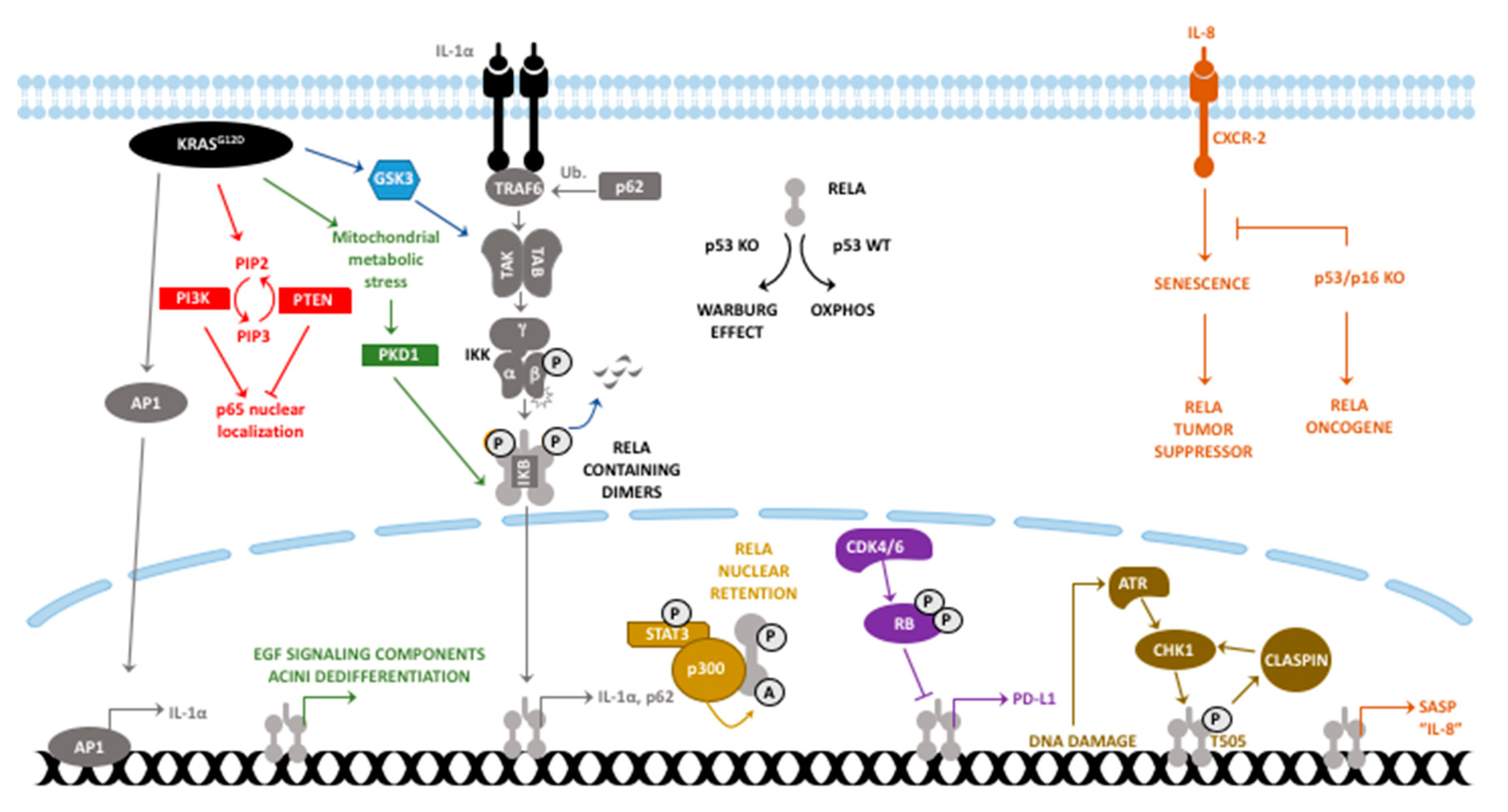
2. RelA/p65 in PDAC
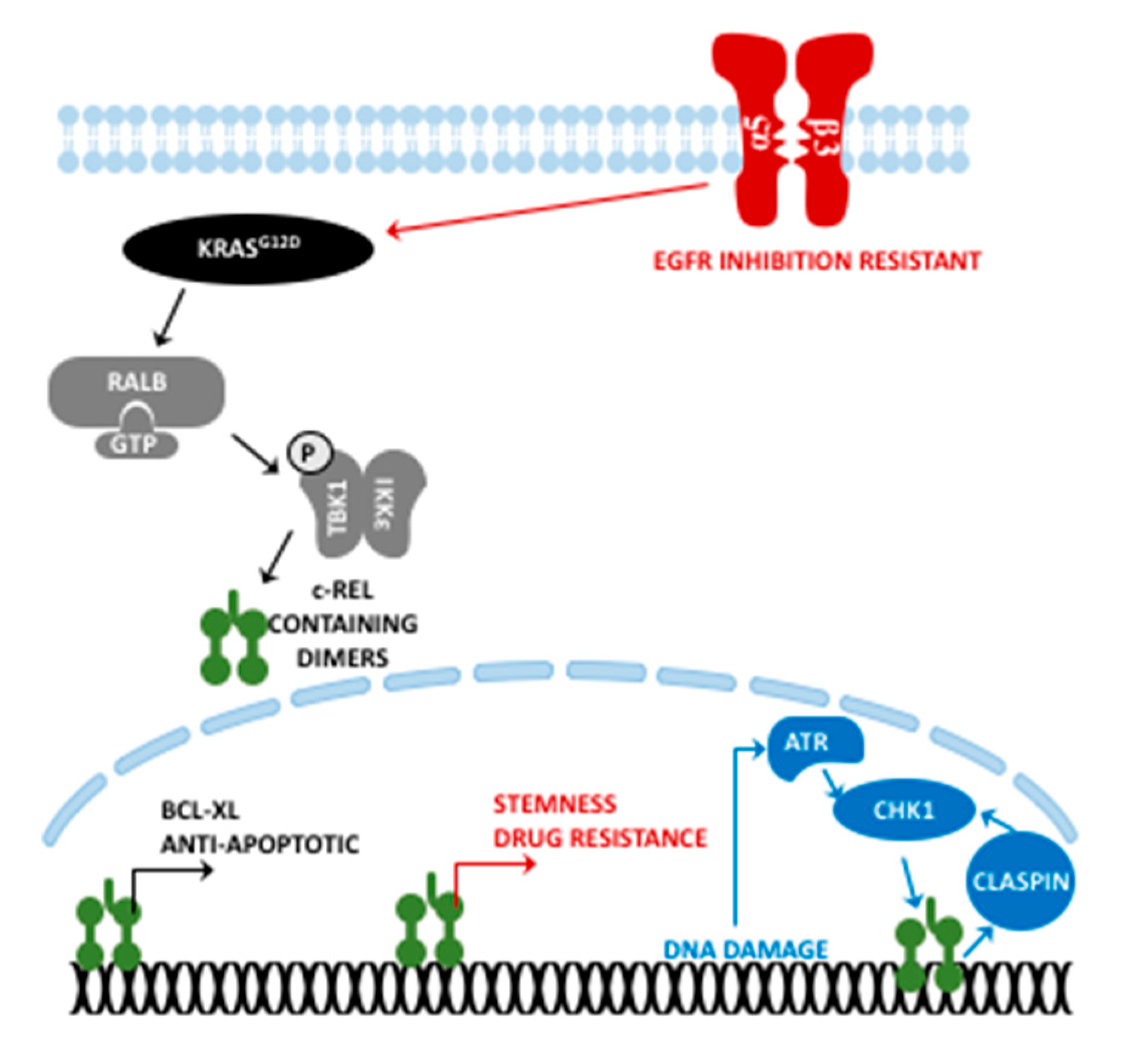
3. c-Rel in PDAC
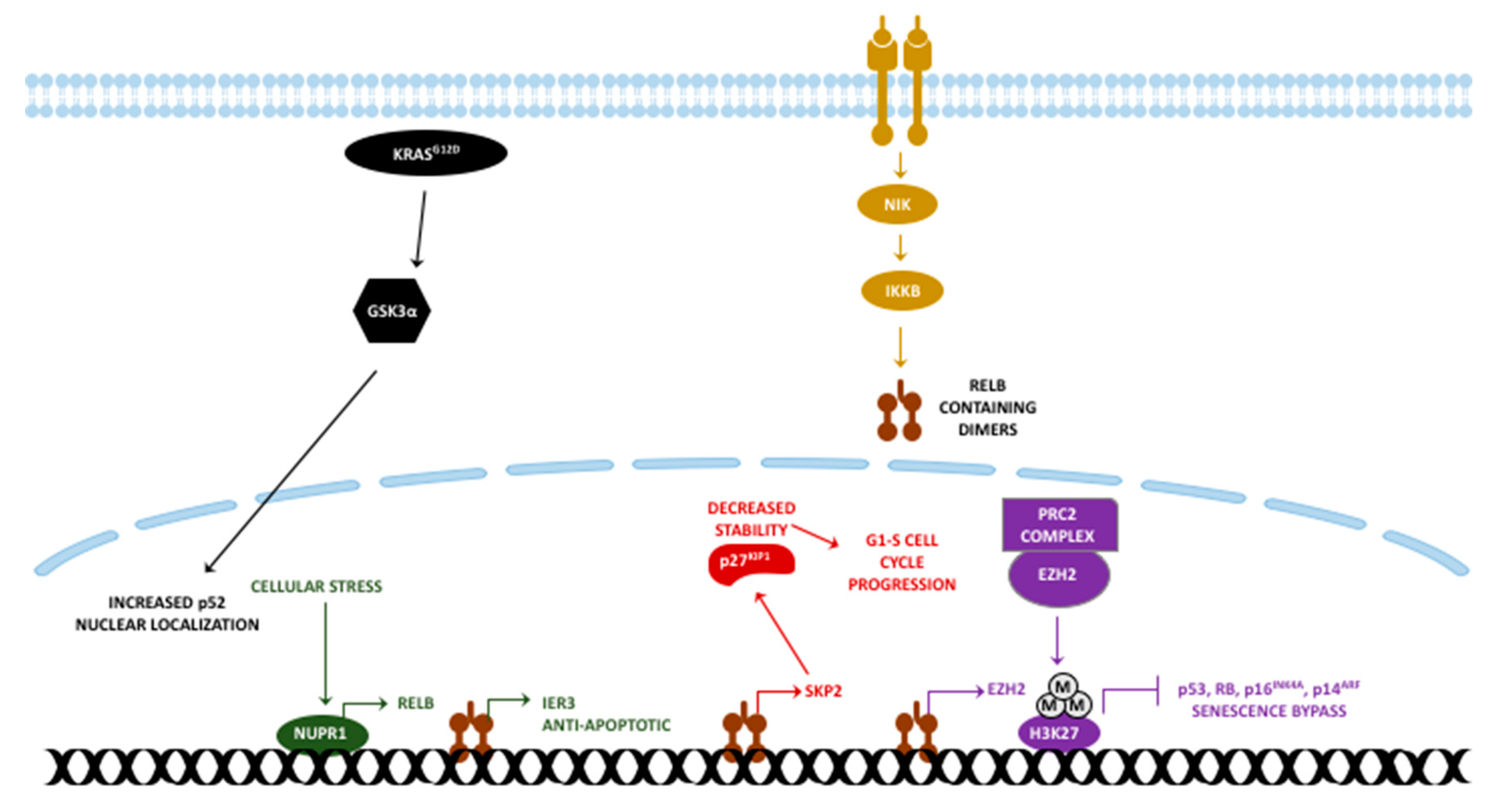
4. RelB in PDAC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Global Cancer Observatory Cancer Tomorrow. Available online: http://gco.iarc.fr/tomorrow/home (accessed on 16 May 2019).
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Ottaiano, A.; Capozzi, M.; Divitiis, C.D.; Stefano, A.D.; Botti, G.; Avallone, A.; Tafuto, S. Gemcitabine mono-therapy versus gemcitabine plus targeted therapy in advanced pancreatic cancer: A meta-analysis of randomized phase III trials. Acta Oncol. 2017, 56, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Brosnan, J.A.; Iacobuzio-Donahue, C.A. Pancreatic cancer genomics: Insights and opportunities for clinical translation. Genome Med. 2013, 5, 26. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Kong, B.; Esposito, I.; Friess, H.; Kleeff, J. The impact of the activated stroma on pancreatic ductal adenocarcinoma biology and therapy resistance. Curr. Mol. Med. 2012, 12, 288–303. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Srinivasan, V.; Kriete, A.; Sacan, A.; Michal Jazwinski, S. Comparing the Yeast Retrograde Response and NF-kB Stress Responses: Implications for Aging. Aging Cell 2010, 9, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Irazoqui, J.E.; Urbach, J.M.; Ausubel, F.M. Evolution of host innate defence: Insights from C. elegans and primitive invertebrates. Nat. Rev. Immunol. 2010, 10, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Wolenski, F.S.; Garbati, M.R.; Lubinski, T.J.; Traylor-Knowles, N.; Dresselhaus, E.; Stefanik, D.J.; Goucher, H.; Finnerty, J.R.; Gilmore, T.D. Characterization of the Core Elements of the NF-κB Signaling Pathway of the Sea Anemone Nematostella vectensis. Mol. Cell. Biol. 2011, 31, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell 1988, 53, 211–217. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Haskill, S.; Beg, A.A.; Tompkins, S.M.; Morris, J.S.; Yurochko, A.D.; Sampson-Johannes, A.; Mondal, K.; Ralph, P.; Baldwin, A.S. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell 1991, 65, 1281–1289. [Google Scholar] [CrossRef]
- Link, E.; Kerr, L.D.; Schreck, R.; Zabel, U.; Verma, I.; Baeuerle, P.A. Purified I kappa B-beta is inactivated upon dephosphorylation. J. Biol. Chem. 1992, 267, 239–246. [Google Scholar]
- Bours, V.; Villalobos, J.; Burd, P.R.; Kelly, K.; Siebenlist, U. Cloning of a mitogen-inducible gene encoding a kappa B DNA-binding protein with homology to the rel oncogene and to cell-cycle motifs. Nature 1990, 348, 76–80. [Google Scholar] [CrossRef]
- Ghosh, S.; Gifford, A.M.; Riviere, L.R.; Tempst, P.; Nolan, G.P.; Baltimore, D. Cloning of the p50 DNA binding subunit of NF-kappa B: Homology to rel and dorsal. Cell 1990, 62, 1019–1029. [Google Scholar] [CrossRef]
- Kieran, M.; Blank, V.; Logeat, F.; Vandekerckhove, J.; Lottspeich, F.; Le Bail, O.; Urban, M.B.; Kourilsky, P.; Baeuerle, P.A.; Israël, A. The DNA binding subunit of NF-kappa B is identical to factor KBF1 and homologous to the rel oncogene product. Cell 1990, 62, 1007–1018. [Google Scholar] [CrossRef]
- Meyer, R.; Hatada, E.N.; Hohmann, H.P.; Haiker, M.; Bartsch, C.; Röthlisberger, U.; Lahm, H.W.; Schlaeger, E.J.; van Loon, A.P.; Scheidereit, C. Cloning of the DNA-binding subunit of human nuclear factor kappa B: The level of its mRNA is strongly regulated by phorbol ester or tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1991, 88, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.M.; Perkins, N.D.; Duckett, C.S.; Andrews, P.C.; Nabel, G.J. Cloning of an NF-kappa B subunit which stimulates HIV transcription in synergy with p65. Nature 1991, 352, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Chang, C.C.; Lombardi, L.; Salina, M.; Corradini, P.; Maiolo, A.T.; Chaganti, R.S.; Dalla-Favera, R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kappa B p50. Cell 1991, 67, 1075–1087. [Google Scholar] [CrossRef]
- Mercurio, F.; Didonato, J.; Rosette, C.; Karin, M. Molecular cloning and characterization of a novel Rel/NF-kappa B family member displaying structural and functional homology to NF-kappa B p50/p105. DNA Cell Biol. 1992, 11, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Burd, P.R.; Brown, K.; Villalobos, J.; Park, S.; Ryseck, R.P.; Bravo, R.; Kelly, K.; Siebenlist, U. A novel mitogen-inducible gene product related to p50/p105-NF-kappa B participates in transactivation through a kappa B site. Mol. Cell. Biol. 1992, 12, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Connelly, M.A.; Marcu, K.B. CHUK, a new member of the helix-loop-helix and leucine zipper families of interacting proteins, contains a serine-threonine kinase catalytic domain. Cell. Mol. Biol. Res. 1995, 41, 537–549. [Google Scholar]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef]
- Mercurio, F.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Young, D.B.; Li, J.W.; Pascual, G.; Motiwala, A.; Zhu, H.; Mann, M.; et al. IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol. Cell. Biol. 1999, 19, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, S.; Courtois, G.; Bessia, C.; Whiteside, S.T.; Weil, R.; Agou, F.; Kirk, H.E.; Kay, R.J.; Israël, A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 1998, 93, 1231–1240. [Google Scholar] [CrossRef]
- Rothwarf, D.M.; Zandi, E.; Natoli, G.; Karin, M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef]
- Sizemore, N.; Lerner, N.; Dombrowski, N.; Sakurai, H.; Stark, G.R. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 2002, 277, 3863–3869. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Russo, M.P.; Sartor, R.B.; Jobin, C. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J. Biol. Chem. 2002, 277, 38168–38178. [Google Scholar] [CrossRef]
- Mattioli, I.; Sebald, A.; Bucher, C.; Charles, R.-P.; Nakano, H.; Doi, T.; Kracht, M.; Schmitz, M.L. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 2004, 172, 6336–6344. [Google Scholar] [CrossRef]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lu, Q.; Bottero, V.; Estepa, G.; Morrison, L.; Mercurio, F.; Verma, I.M. Enhanced NF-κB activation and cellular function in macrophages lacking IκB kinase 1 (IKK1). Proc. Natl. Acad. Sci. USA 2005, 102, 12425–12430. [Google Scholar] [CrossRef] [PubMed]
- Pradère, J.-P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-κB p65 activity by serine 536 phosphorylation. Sci. Signal. 2016, 9, ra85. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational Biogenesis of NF-κB p50 by the 26S Proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.-C.; et al. Activation by IKKα of a Second, Evolutionary Conserved, NF-κB Signaling Pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef]
- Coope, H.J.; Atkinson, P.G.P.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.J.; Klaus, G.G.B.; Johnston, L.H.; Ley, S.C. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.-W.; Karin, M.; Ware, C.F.; Green, D.R. The Lymphotoxin-β Receptor Induces Different Patterns of Gene Expression via Two NF-κB Pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Novack, D.V.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S.L. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef]
- Fong, A.; Sun, S.-C. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J. Biol. Chem. 2002, 277, 22111–22114. [Google Scholar] [CrossRef]
- Xiao, G.; Fong, A.; Sun, S.-C. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [PubMed]
- Bours, V.; Azarenko, V.; Dejardin, E.; Siebenlist, U. Human RelB (I-Rel) functions as a kappa B site-dependent transactivating member of the family of Rel-related proteins. Oncogene 1994, 9, 1699–1702. [Google Scholar] [PubMed]
- Arenzana-Seisdedos, F.; Thompson, J.; Rodriguez, M.S.; Bachelerie, F.; Thomas, D.; Hay, R.T. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol. Cell. Biol. 1995, 15, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Arenzana-Seisdedos, F.; Turpin, P.; Rodriguez, M.; Thomas, D.; Hay, R.T.; Virelizier, J.L.; Dargemont, C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell. Sci. 1997, 110 Pt 3, 369–378. [Google Scholar] [PubMed]
- Hong, J.-W.; Allen, C.E.; Wu, L.-C. Inhibition of NF-κB by ZAS3, a zinc-finger protein that also binds to the κB motif. Proc. Natl. Acad. Sci. USA 2003, 100, 12301–12306. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.D.; Yoon, K.; Shin, Y.J.; Kim, J.; Lee, S.Y. PIAS3 Suppresses NF-κB-mediated Transcription by Interacting with the p65/RelA Subunit. J. Biol. Chem. 2004, 279, 24873–24880. [Google Scholar] [CrossRef]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Frank, R.; Livingstone, M.; Resch, K.; Kracht, M. Phosphorylation of Serine 468 by GSK-3β Negatively Regulates Basal p65 NF-κB Activity. J. Biol. Chem. 2004, 279, 49571–49574. [Google Scholar] [CrossRef]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Cheng, C.S.; Feldman, K.E.; Lee, J.; Verma, S.; Huang, D.-B.; Huynh, K.; Chang, M.; Ponomarenko, J.V.; Sun, S.-C.; Benedict, C.A.; et al. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci. Signal. 2011, 4, ra11. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.Y.-F.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The Transcriptional Specificity of NF-κB Dimers Is Coded within the κB DNA Response Elements. Cell Rep. 2012, 2, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; de Jesus, T.; Shukla, S. NF-kB c-Rel Dictates the Inflammatory Threshold by Acting as a Transcriptional Repressor. J. Immunol. 2018, 200, 109-9. [Google Scholar]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer: NF-κB links inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Niu, J.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Inhibition of constitutive NF-κB activity by IκBαM suppresses tumorigenesis. Oncogene 2003, 22, 1365–1370. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-Induced IKK2/β/NF-κB Activation by IL-1α and p62 Feedforward Loops Is Required for Development of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Maier, H.J.; Wagner, M.; Schips, T.G.; Salem, H.H.; Baumann, B.; Wirth, T. Requirement of NEMO/IKKγ for effective expansion of KRAS-induced precancerous lesions in the pancreas. Oncogene 2013, 32, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Polykratis, A.; Ehlken, H.; Ochoa-Callejero, L.; Straub, B.K.; Krishna-Subramanian, S.; Van, T.-M.; Curth, H.-M.; Heise, N.; Weih, F.; et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell 2015, 28, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.L.; Hawke, N.A.; Kashatus, D.; Baldwin, A.S. The nuclear factor kappaB subunits RelA/p65 and c-Rel potentiate but are not required for Ras-induced cellular transformation. Cancer Res. 2004, 64, 7248–7255. [Google Scholar] [CrossRef] [PubMed]
- Finco, T.S.; Westwick, J.K.; Norris, J.L.; Beg, A.A.; Der, C.J.; Baldwin, A.S. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J. Biol. Chem. 1997, 272, 24113–24116. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-κB Subunit p65/RelA for K-Ras–Induced Lung Tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar] [PubMed]
- Lallena, M.-J.; Diaz-Meco, M.T.; Bren, G.; Payá, C.V.; Moscat, J. Activation of IκB Kinase β by Protein Kinase C Isoforms. Mol. Cell. Biol. 1999, 19, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 Scaffold Regulates Nerve Growth Factor-induced NF-κB Activation by Influencing TRAF6 Polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Diaz-Meco, M.T.; Moscat, J. The signaling adapter p62 is an important mediator of T helper 2 cell function and allergic airway inflammation. EMBO J. 2006, 25, 3524–3533. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The Signaling Adaptor p62 Is an Important NF-κB Mediator in Tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-κB pathway–mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Gurumurthy, S.; Rangnekar, V.M. Suppression of PTEN Expression by NF-B Prevents Apoptosis. Mol. Cell. Biol. 2004, 24, 1007–1021. [Google Scholar] [CrossRef]
- Wilson, W.; Baldwin, A.S. Maintenance of Constitutive I B Kinase Activity by Glycogen Synthase Kinase-3/in Pancreatic Cancer. Cancer Res. 2008, 68, 8156–8163. [Google Scholar] [CrossRef]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α Promotes Oncogenic KRAS Function in Pancreatic Cancer via TAK1–TAB Stabilization and Regulation of Noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef]
- Lu, T.; Sathe, S.S.; Swiatkowski, S.M.; Hampole, C.V.; Stark, G.R. Secretion of cytokines and growth factors as a general cause of constitutive NF κ B activation in cancer. Oncogene 2004, 23, 2138. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Wang, S.C.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated with Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.-H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Loss of p53 enhances catalytic activity of IKKβ through O-linked β-N-acetyl glucosamine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 3431–3436. [Google Scholar] [CrossRef]
- Johnson, R.F.; Witzel, I.-I.; Perkins, N.D. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-κB. Cancer Res. 2011, 71, 5588–5597. [Google Scholar] [CrossRef]
- Cogswell, P.C.; Kashatus, D.F.; Keifer, J.A.; Guttridge, D.C.; Reuther, J.Y.; Bristow, C.; Roy, S.; Nicholson, D.W.; Baldwin, A.S. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J. Biol. Chem. 2003, 278, 2963–2968. [Google Scholar] [CrossRef]
- Guseva, N.V.; Taghiyev, A.F.; Sturm, M.T.; Rokhlin, O.W.; Cohen, M.B. Tumor necrosis factor-related apoptosis-inducing ligand-mediated activation of mitochondria-associated nuclear factor-kappaB in prostatic carcinoma cell lines. Mol. Cancer Res. 2004, 2, 574–584. [Google Scholar]
- Algül, H.; Treiber, M.; Lesina, M.; Nakhai, H.; Saur, D.; Geisler, F.; Pfeifer, A.; Paxian, S.; Schmid, R.M. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J. Clin. Investig. 2007, 117, 1490–1501. [Google Scholar] [CrossRef]
- Lesina, M.; Wörmann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwächter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 2016, 126, 2919–2932. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Wang, J.; Jacob, N.K.; Ladner, K.J.; Beg, A.; Perko, J.D.; Tanner, S.M.; Liyanarachchi, S.; Fishel, R.; Guttridge, D.C. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009, 10, 1272–1278. [Google Scholar] [CrossRef]
- Caldwell, M.E.; DeNicola, G.M.; Martins, C.P.; Jacobetz, M.A.; Maitra, A.; Hruban, R.H.; Tuveson, D.A. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012, 31, 1599–1608. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signaling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Lee, K.E.; Bar-Sagi, D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 2010, 18, 448–458. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-κB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e6. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Rocha, S.; Garrett, M.D.; Campbell, K.J.; Schumm, K.; Perkins, N.D. Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005, 24, 1157–1169. [Google Scholar] [CrossRef]
- Moles, A.; Butterworth, J.A.; Sanchez, A.; Hunter, J.E.; Leslie, J.; Sellier, H.; Tiniakos, D.; Cockell, S.J.; Mann, D.A.; Oakley, F.; et al. A RelA(p65) Thr505 phospho-site mutation reveals an important mechanism regulating NF-κB-dependent liver regeneration and cancer. Oncogene 2016, 35, 4623–4632. [Google Scholar] [CrossRef]
- Regulation of Checkpoint Kinase Signalling and Tumorigenesis by the NF-κB Regulated Gene, CLSPN|bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/358291v1 (accessed on 20 May 2019).
- Campbell, K.J.; Rocha, S.; Perkins, N.D. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol. Cell 2004, 13, 853–865. [Google Scholar] [CrossRef]
- Campbell, K.J.; Witty, J.M.; Rocha, S.; Perkins, N.D. Cisplatin Mimics ARF Tumor Suppressor Regulation of RelA (p65) Nuclear Factor-κB Transactivation. Cancer Res. 2006, 66, 929–935. [Google Scholar] [CrossRef]
- Sabatel, H.; Valentin, E.D.; Gloire, G.; Dequiedt, F.; Piette, J.; Habraken, Y. Phosphorylation of p65(RelA) on Ser547 by ATM Represses NF-κB-Dependent Transcription of Specific Genes after Genotoxic Stress. PLoS ONE 2012, 7, e38246. [Google Scholar] [CrossRef]
- Kong, R.; Sun, B.; Jiang, H.; Pan, S.; Chen, H.; Wang, S.; Krissansen, G.W.; Sun, X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010, 291, 90–98. [Google Scholar] [CrossRef]
- Zhuang, Z.; Ju, H.-Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-κB Activation. Clin. Cancer Res. 2016, 22, 1432–1444. [Google Scholar] [CrossRef]
- Skrypek, N.; Duchêne, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef]
- Mezencev, R.; Matyunina, L.V.; Wagner, G.T.; McDonald, J.F. Acquired resistance of pancreatic cancer cells to cisplatin is multifactorial with cell context-dependent involvement of resistance genes. Cancer Gene Ther. 2016, 23, 446–453. [Google Scholar] [CrossRef]
- Simek, S.; Rice, N.R. Detection and characterization of the protein encoded by the chicken c-rel protooncogene. Oncogene Res. 1988, 2, 103–119. [Google Scholar]
- Lim, M.Y.; Davis, N.; Zhang, J.Y.; Bose, H.R. The v-rel oncogene product is complexed with cellular proteins including its proto-oncogene product and heat shock protein 70. Virology 1990, 175, 149–160. [Google Scholar] [CrossRef]
- Fan, Y.; Rayet, B.; Gélinas, C. Divergent C-terminal transactivation domains of Rel/NF-κB proteins are critical determinants of their oncogenic potential in lymphocytes. Oncogene 2004, 23, 1030. [Google Scholar] [CrossRef][Green Version]
- Romieu-Mourez, R.; Kim, D.W.; Shin, S.M.; Demicco, E.G.; Landesman-Bollag, E.; Seldin, D.C.; Cardiff, R.D.; Sonenshein, G.E. Mouse Mammary Tumor Virus c-rel Transgenic Mice Develop Mammary Tumors. Mol. Cell. Biol. 2003, 23, 5738–5754. [Google Scholar] [CrossRef]
- Belguise, K.; Sonenshein, G.E. PKCtheta promotes c-Rel-driven mammary tumorigenesis in mice and humans by repressing estrogen receptor alpha synthesis. J. Clin. Investig. 2007, 117, 4009–4021. [Google Scholar]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef]
- Kunsch, C.; Ruben, S.M.; Rosen, C.A. Selection of optimal kappa B/Rel DNA-binding motifs: Interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 4412–4421. [Google Scholar] [CrossRef]
- Hunter, J.E.; Leslie, J.; Perkins, N.D. c-Rel and its many roles in cancer: An old story with new twists. Br. J. Cancer 2016, 114, 1–6. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.-M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M.; et al. RalB GTPase-Mediated Activation of the IκB Family Kinase TBK1 Couples Innate Immune Signaling to Tumor Cell Survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef]
- Harris, J.; Olière, S.; Sharma, S.; Sun, Q.; Lin, R.; Hiscott, J.; Grandvaux, N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKK epsilon. J. Immunol. 2006, 177, 2527–2535. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef]
- Muvaffak, A.; Pan, Q.; Yan, H.; Fernandez, R.; Lim, J.; Dolinski, B.; Nguyen, T.T.; Strack, P.; Wu, S.; Chung, R.; et al. Evaluating TBK1 as a therapeutic target in cancers with activated IRF3. Mol. Cancer Res. 2014, 12, 1055–1066. [Google Scholar] [CrossRef]
- Ou, Y.-H.; Torres, M.; Ram, R.; Formstecher, E.; Roland, C.; Cheng, T.; Brekken, R.; Wurz, R.; Tasker, A.; Polverino, T.; et al. TBK1 Directly Engages Akt/PKB Survival Signaling to Support Oncogenic Transformation. Mol. Cell 2011, 41, 458–470. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Mudie, S.; Rocha, S. IKK and NF-kappaB-mediated regulation of Claspin impacts on ATR checkpoint function. EMBO J. 2010, 29, 2966–2978. [Google Scholar] [CrossRef]
- Isomura, I.; Palmer, S.; Grumont, R.J.; Bunting, K.; Hoyne, G.; Wilkinson, N.; Banerjee, A.; Proietto, A.; Gugasyan, R.; Wu, L.; et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 2009, 206, 3001–3014. [Google Scholar] [CrossRef]
- Long, M.; Park, S.-G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.-C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development Of Foxp3+ Regulatory T Cells Is Driven By A c-Rel Enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-κB transcription factor-dependent, lineage specific transcriptional program promotes regulatory T cell identity and function. Immunity 2017, 47, 450–465.e5. [Google Scholar] [CrossRef]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef]
- Köntgen, F.; Grumont, R.J.; Strasser, A.; Metcalf, D.; Li, R.; Tarlinton, D.; Gerondakis, S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995, 9, 1965–1977. [Google Scholar] [CrossRef]
- Wang, W.; Tam, W.F.; Hughes, C.C.; Rath, S.; Sen, R. c-Rel is a target of pentoxifylline-mediated inhibition of T lymphocyte activation. Immunity 1997, 6, 165–174. [Google Scholar] [CrossRef]
- Shono, Y.; Tuckett, A.Z.; Ouk, S.; Liou, H.-C.; Altan-Bonnet, G.; Tsai, J.J.; Oyler, J.E.; Smith, O.M.; West, M.L.; Singer, N.V.; et al. A small molecule c-Rel inhibitor reduces alloactivation of T-cells without compromising anti-tumor activity. Cancer Discov. 2014, 4, 578–591. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Hayden, M.S.; Klein, U.; Ghosh, S. NF-κB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e13. [Google Scholar] [CrossRef]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Bengsch, F.; Knoblock, D.M.; Liu, A.; McAllister, F.; Beatty, G.L. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017, 66, 1609–1617. [Google Scholar] [CrossRef]
- Marienfeld, R.; May, M.J.; Berberich, I.; Serfling, E.; Ghosh, S.; Neumann, M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003, 278, 19852–19860. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Petrovas, C.; Sonenshein, G.E. RelB-p50 NF-κB Complexes Are Selectively Induced by Cytomegalovirus Immediate-Early Protein 1: Differential Regulation of Bcl-xL Promoter Activity by NF-κB Family Members. J. Virol. 2002, 76, 5737–5747. [Google Scholar] [CrossRef]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Korner, M.; Baud, V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef]
- Jacque, E.; Tchenio, T.; Piton, G.; Romeo, P.-H.; Baud, V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 14635–14640. [Google Scholar] [CrossRef]
- Ryseck, R.P.; Bull, P.; Takamiya, M.; Bours, V.; Siebenlist, U.; Dobrzanski, P.; Bravo, R. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol. Cell. Biol. 1992, 12, 674–684. [Google Scholar] [CrossRef]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Wharry, C.E.; Haines, K.M.; Carroll, R.G.; May, M.J. Constitutive non-canonical NFkappaB signaling in pancreatic cancer cells. Cancer Biol. Ther. 2009, 8, 1567–1576. [Google Scholar] [CrossRef]
- Döppler, H.; Liou, G.-Y.; Storz, P. Downregulation of TRAF2 Mediates NIK-Induced Pancreatic Cancer Cell Proliferation and Tumorigenicity. PLoS ONE 2013, 8, e53676. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Hamidi, T.; Algül, H.; Cano, C.E.; Sandi, M.J.; Molejon, M.I.; Riemann, M.; Calvo, E.L.; Lomberk, G.; Dagorn, J.-C.; Weih, F.; et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J. Clin. Investig. 2012, 122, 2092–2103. [Google Scholar] [CrossRef]
- Grasso, D.; Bintz, J.; Lomberk, G.; Molejon, M.I.; Loncle, C.; Garcia, M.N.; Lopez, M.B.; Urrutia, R.; Iovanna, J.L. Pivotal Role of the Chromatin Protein Nupr1 in Kras-Induced Senescence and Transformation. Sci. Rep. 2015, 5, 17549. [Google Scholar] [CrossRef]
- Schneider, G.; Saur, D.; Siveke, J.T.; Fritsch, R.; Greten, F.R.; Schmid, R.M. IKKα controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. EMBO J. 2006, 25, 3801–3812. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Iannetti, A.; Ledoux, A.C.; Tudhope, S.J.; Sellier, H.; Zhao, B.; Mowla, S.; Moore, A.; Hummerich, H.; Gewurz, B.E.; Cockell, S.J.; et al. Regulation of p53 and Rb links the alternative NF-κB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014, 10, e1004642. [Google Scholar] [CrossRef]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.-C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- De Donatis, G.M.; Le Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S.; et al. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2735–2745. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- Shain, A.H.; Giacomini, C.P.; Matsukuma, K.; Karikari, C.A.; Bashyam, M.D.; Hidalgo, M.; Maitra, A.; Pollack, J.R. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E252–E259. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Wang, S.C.; Nassour, I.; Xiao, S.; Zhang, S.; Luo, X.; Lee, J.; Li, L.; Sun, X.; Nguyen, L.H.; Chuang, J.-C.; et al. SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 2018. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Hou, Y.; Liang, H.; Rao, E.; Zheng, W.; Huang, X.; Deng, L.; Zhang, Y.; Yu, X.; Xu, M.; Mauceri, H.; et al. Non-canonical NF-κB Antagonizes STING Sensor-Mediated DNA Sensing in Radiotherapy. Immunity 2018, 49, 490–503.e4. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Q.; Dudek, A.Z. Phase II Study of Panobinostat and Bortezomib in Patients with Pancreatic Cancer Progressing on Gemcitabine-based Therapy. Anticancer Res. 2012, 32, 1027–1031. [Google Scholar] [PubMed]
- Dudek, A.Z.; Lesniewski-Kmak, K.; Shehadeh, N.J.; Pandey, O.N.; Franklin, M.; Kratzke, R.A.; Greeno, E.W.; Kumar, P. Phase I study of bortezomib and cetuximab in patients with solid tumours expressing epidermal growth factor receptor. Br. J. Cancer 2009, 100, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Bekaii-Saab, T.; Schaaf, L.J.; Lesinski, G.B.; Lucas, D.M.; Young, D.C.; Ruppert, A.S.; Byrd, J.C.; Culler, K.; Wilkins, D.; et al. A dose-finding and pharmacodynamic study of bortezomib in combination with weekly paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2010, 66, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Epelbaum, R.; Schaffer, M.; Vizel, B.; Badmaev, V.; Bar-Sela, G. Curcumin and Gemcitabine in Patients With Advanced Pancreatic Cancer. Nutr. Cancer 2010, 62, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Becerra, C.; Paulson, A.S.; Cavaness, K.M.; Celinski, S.A. Gemcitabine, nab-paclitaxel, cisplatin, and anakinra (AGAP) treatment in patients with localized pancreatic ductal adenocarcinoma (PDAC). JCO 2018, 36, 449. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| NCT Number | Intervention | Disease | Phase | Status | Ref. |
|---|---|---|---|---|---|
| NCT01056601 | Bortezomib (Proteasome/NF-κB Inhibitor) + Panobinostat (HDACi) | Pancreatic cancer progressive upon gemcitabine treatment | II | Terminated: Toxicity and lack of response. | [183] |
| NCT00416793 | Bortezomib (Proteasome/NF-κB inhibitor) + Carboplatin (chemotherapy) | Metastatic pancreatic cancer | II | Terminated: Toxicity and lack of response | n.a. |
| NCT00052689 | Bortezomib (Proteasome/NF-κB inhibitor) ± Gemcitabine (chemotherapy) | Stage IV pancreatic cancer | II | Completed: Results n.a. | n.a. |
| NCT00622674 | Bortezomib (Proteasome/NF-κB inhibitor) + Cetuximab (EGFR inhibitor) | EGFR-expressing solid tumors; 3/37 patients with pancreatic tumor | I | Completed: Treatment dose is tolerable; no response observed in pancreatic cancer | [184] |
| NCT00052689 | Bortezomib (Proteasome/NF-κB inhibitor) ± Gemcitabine (chemotherapy) | Older patients with advanced pancreatic cancer | II | Completed: Results n.a. | n.a. |
| n.a. | Bortezomib (Proteasome/NF-κB inhibitor) + Paclitaxel (chemotherapy) | Advanced solid tumors | I | Completed: Manageable toxicity profile; 7/45 patients showed disease stabilization; 3 had metastatic pancreatic cancer | [185] |
| NCT03878524 | Various targeted/chemotherapy drugs in combination, among them Bortezomib (Proteasome/NF-κB inhibitor) | Advanced cancers including pancreatic | I | Not yet recruiting. The study aims at molecular stratification and combination treatment in a personalized approach. | n.a. |
| NCT00720785 | Autologous, ex vivo expanded NK cells ± Bortezomib (Proteasome/NF-κB inhibitor) | Various cancers including metastatic pancreatic adenocarcinoma | I | Recruiting | n.a. |
| NCT00094445 | Curcumin (Pleiotropic signaling modulator/NF-κB inhibitor) | Advanced pancreatic cancer; no concomitant chemo/radiotherapy | II | Completed: no toxicity; 2/21 patients showed biological activity. | [186] |
| NCT00192842 | Curcumin (Pleiotropic signaling modulator/NF-κB inhibitor) + Gemcitabine (chemotherapy) | Advanced pancreatic cancer | II | Completed: low compliance for high dose oral curcumin in combination with gemcitabine; 1/11 with partial response, 4/11 with stable disease | [187] |
| NCT02336087 | Gemcitabine + nab-Paclitaxel (Chemotherapy) + Metformin + Dietary supplement including curcumin | Unresectable pancreatic cancer | I | Recruiting | n.a. |
| NCT03382340 | IMX-110: nanoparticle encapsulating curcumin and low-dose doxorubicin (chemotherapy) | Advanced solid tumors | I/II | Recruiting | n.a. |
| NCT02671890 | Gemcitabine (chemotherapy) ± Disulfiram (Proteasome/NF-κB inhibitor) | Unresectable solid tumors or metastatic pancreatic cancer | I | Recruiting | n.a. |
| NCT02550327 | Gemcitabine, Nab-Paclitaxel, Cisplatin and Anakinra (IL-1R antagonist) | Localized pancreatic ductal adenocarcinoma | Early I | Completed: Combination is tolerable. Further analysis pending | [188] |
| NCT01632306 | LY2090314 (GSK-3 inhibitor) in combination with various chemotherapy regimens (Gemcitabine, FOLFOX, nab-Paclitaxel) | Metastatic pancreatic cancer | I/II | Terminated: Lack of patient enrollment. | n.a. |
| NCT03678883 | 9-ING-41 (GSK-3β inhibitor) ± various chemotherapy regimens | Advanced cancers including pancreatic | I/II | Recruiting | n.a. |
| NCT03454035 | Palbociclib (CDK4/6 inhibitor) Ulixertinib (ERK1/2 inhibitor) | Advanced pancreatic cancer and other solid tumors | I | Recruiting | n.a. |
| NCT02465060 | Various targeted therapies including Palbociclib (CDK4/6 inhibitor) BAY 80-6946 (PI3K inhibitor) | Multiple tumor types including refractory pancreatic cancer, treatment option to be evaluated based on genetic testing | II | Recruiting | n.a. |
| NCT03065062 | Palbociclib (CDK4/6 inhibitor) Gedatolisib (PI3K/mTOR inhibitor) | Various solid tumors including advanced pancreatic cancer | I | Recruiting | n.a. |
| NCT03682289 | AZD6738 (ATR inhibitor) ± Olaparib (PARP inhibitor) | Various solid tumors including advanced pancreatic cancer | II | Recruiting | n.a. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabacaoglu, D.; Ruess, D.A.; Ai, J.; Algül, H. NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers 2019, 11, 937. https://doi.org/10.3390/cancers11070937
Kabacaoglu D, Ruess DA, Ai J, Algül H. NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers. 2019; 11(7):937. https://doi.org/10.3390/cancers11070937
Chicago/Turabian StyleKabacaoglu, Derya, Dietrich A. Ruess, Jiaoyu Ai, and Hana Algül. 2019. "NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB" Cancers 11, no. 7: 937. https://doi.org/10.3390/cancers11070937
APA StyleKabacaoglu, D., Ruess, D. A., Ai, J., & Algül, H. (2019). NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers, 11(7), 937. https://doi.org/10.3390/cancers11070937