Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
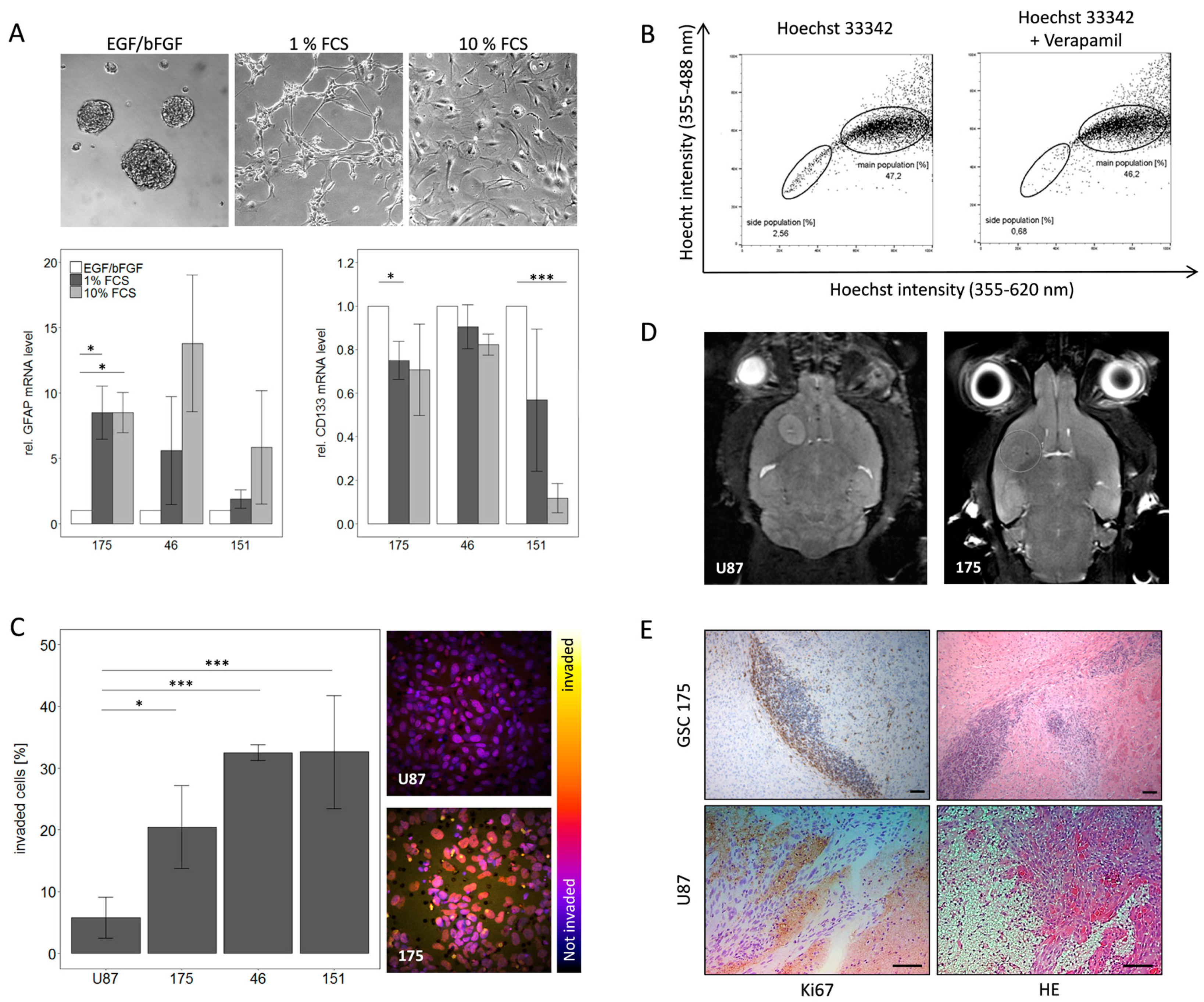
2.1. Characterization of Primary GBM Stem-Like Cells Compared to the Established Cell Line U87
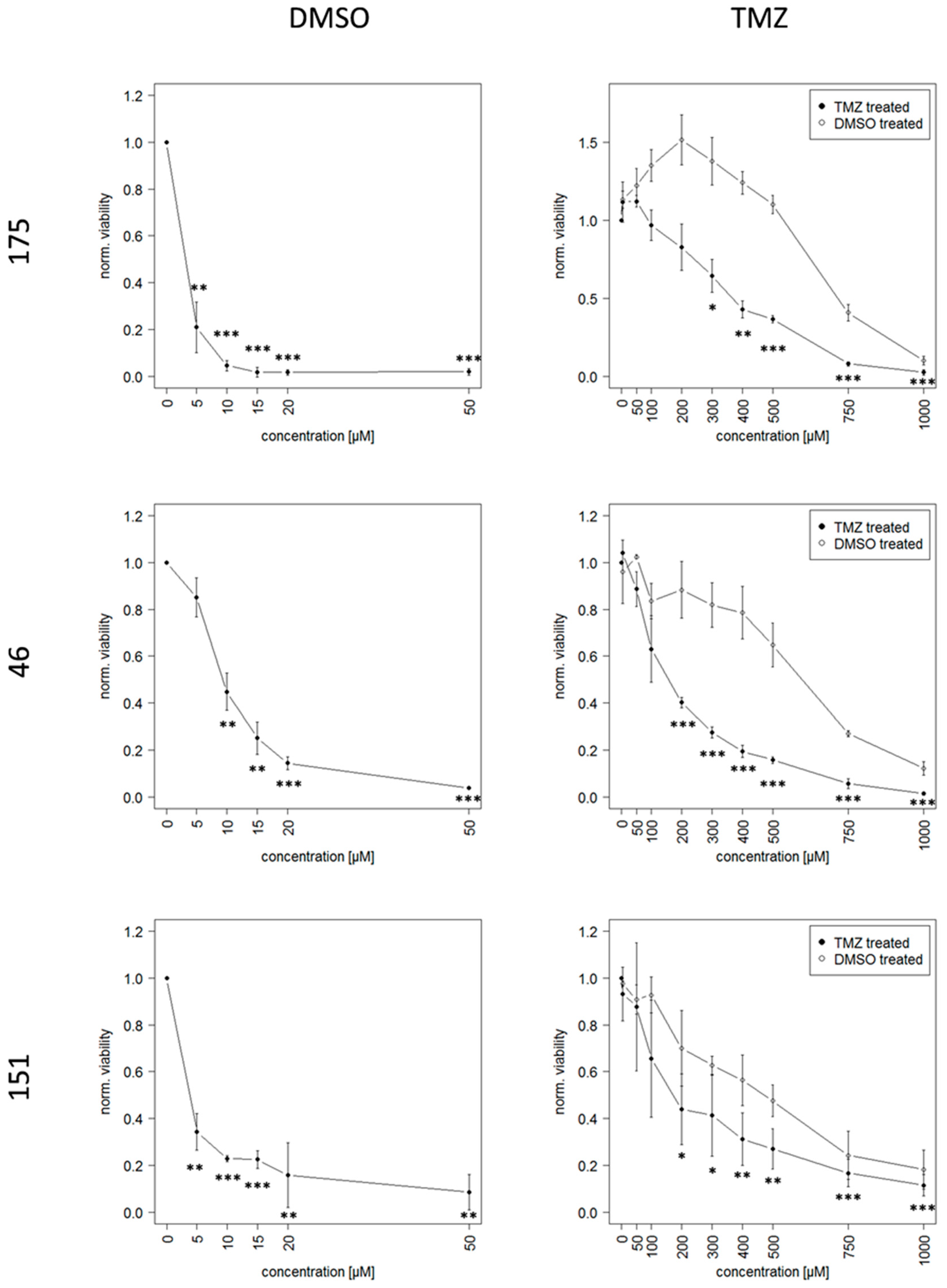
2.2. Long Term Exposure to TMZ Generates Resistant Cells as An In Vitro Model of Reccurence
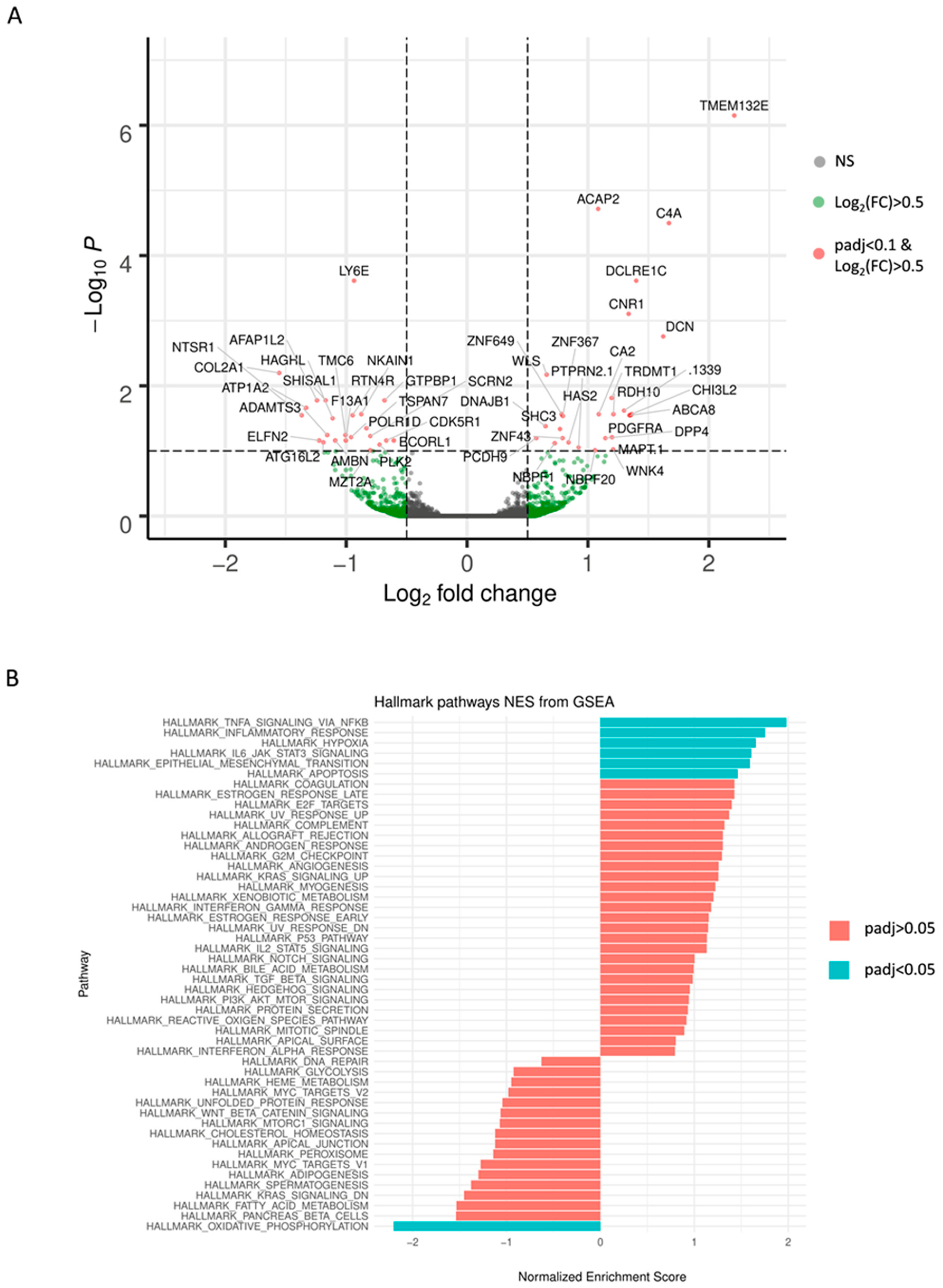
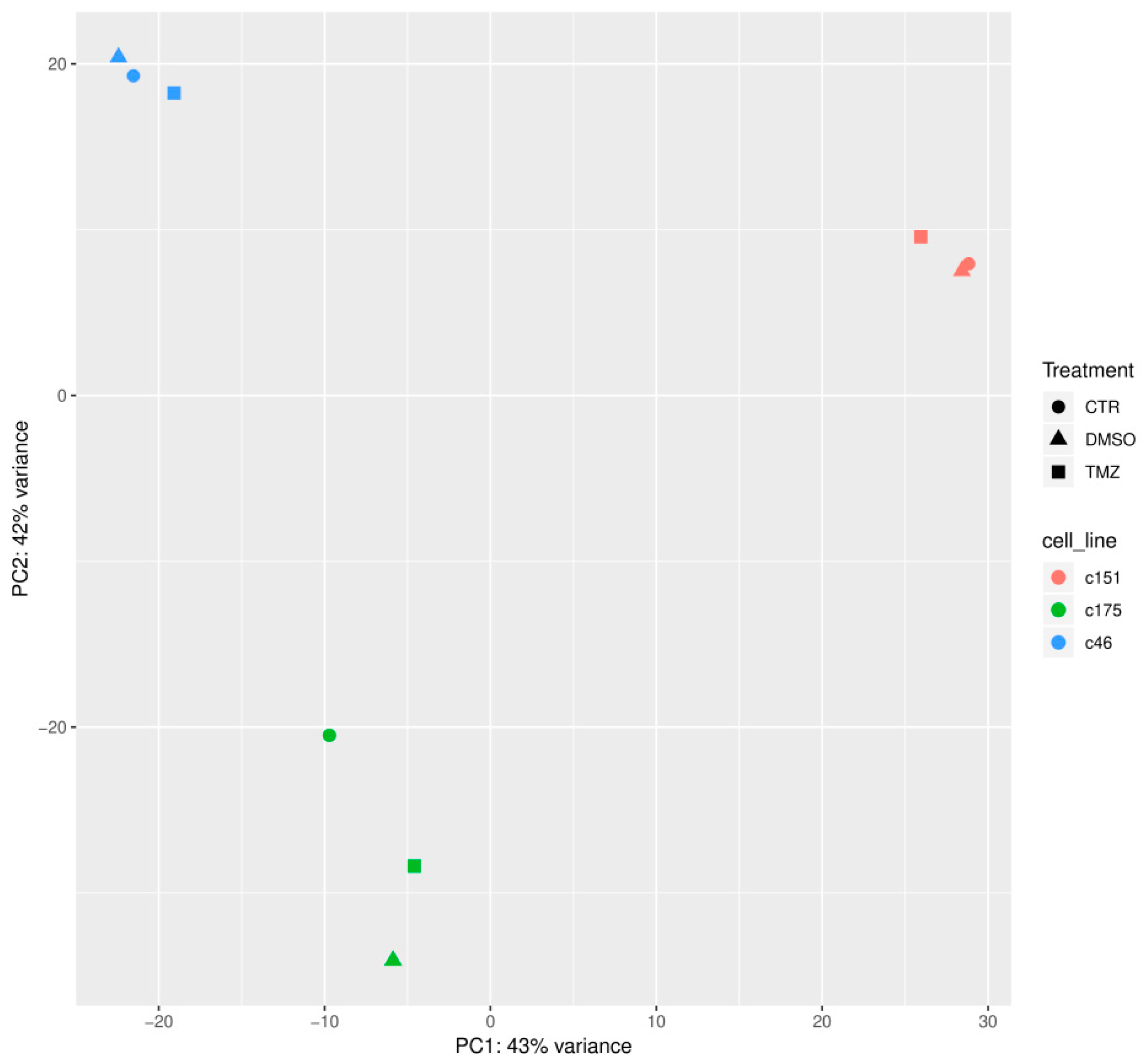
2.3. Identification of Genetic Alterations Causing TMZ Resistance by RNA Sequencing Analysis
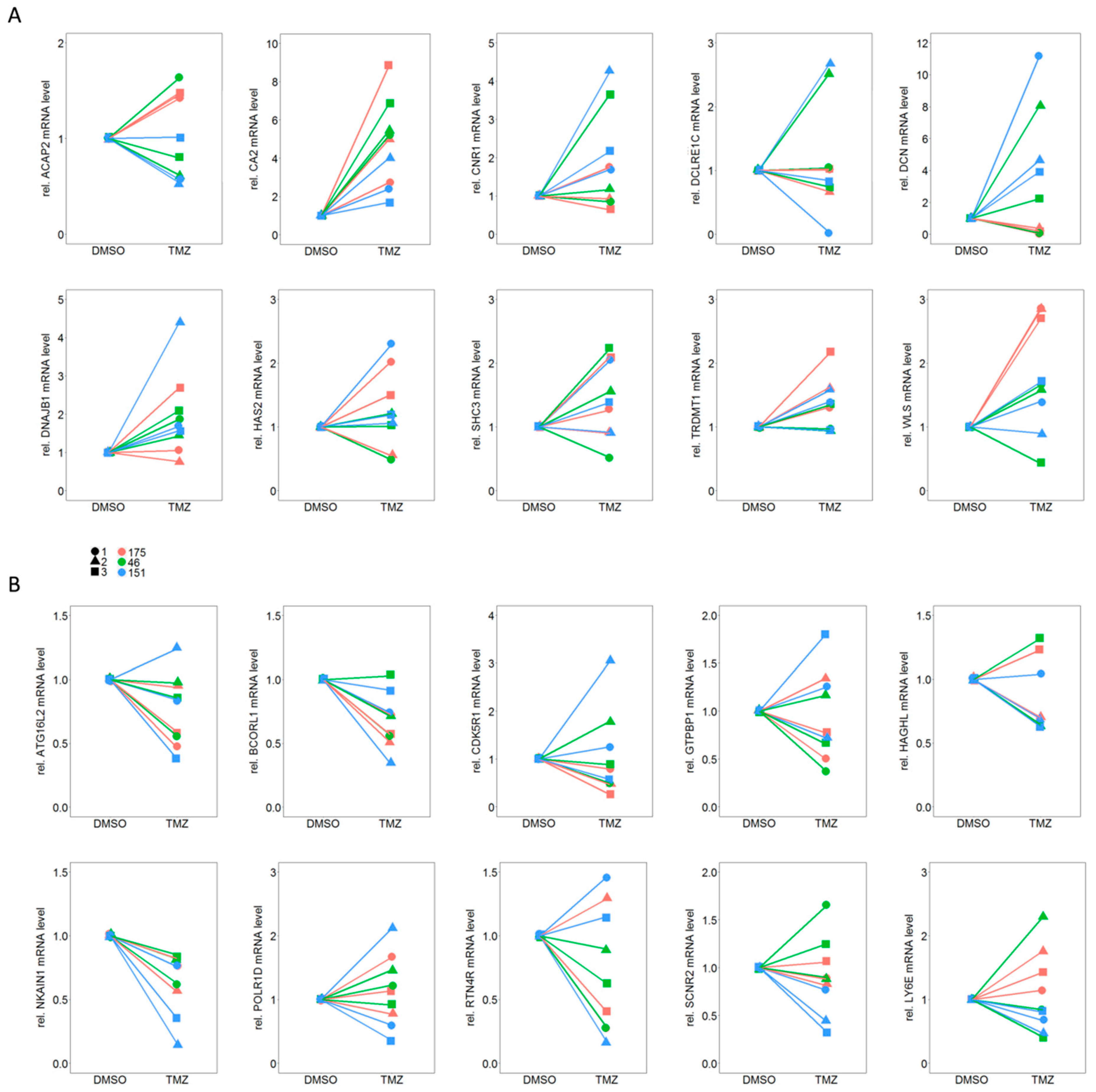
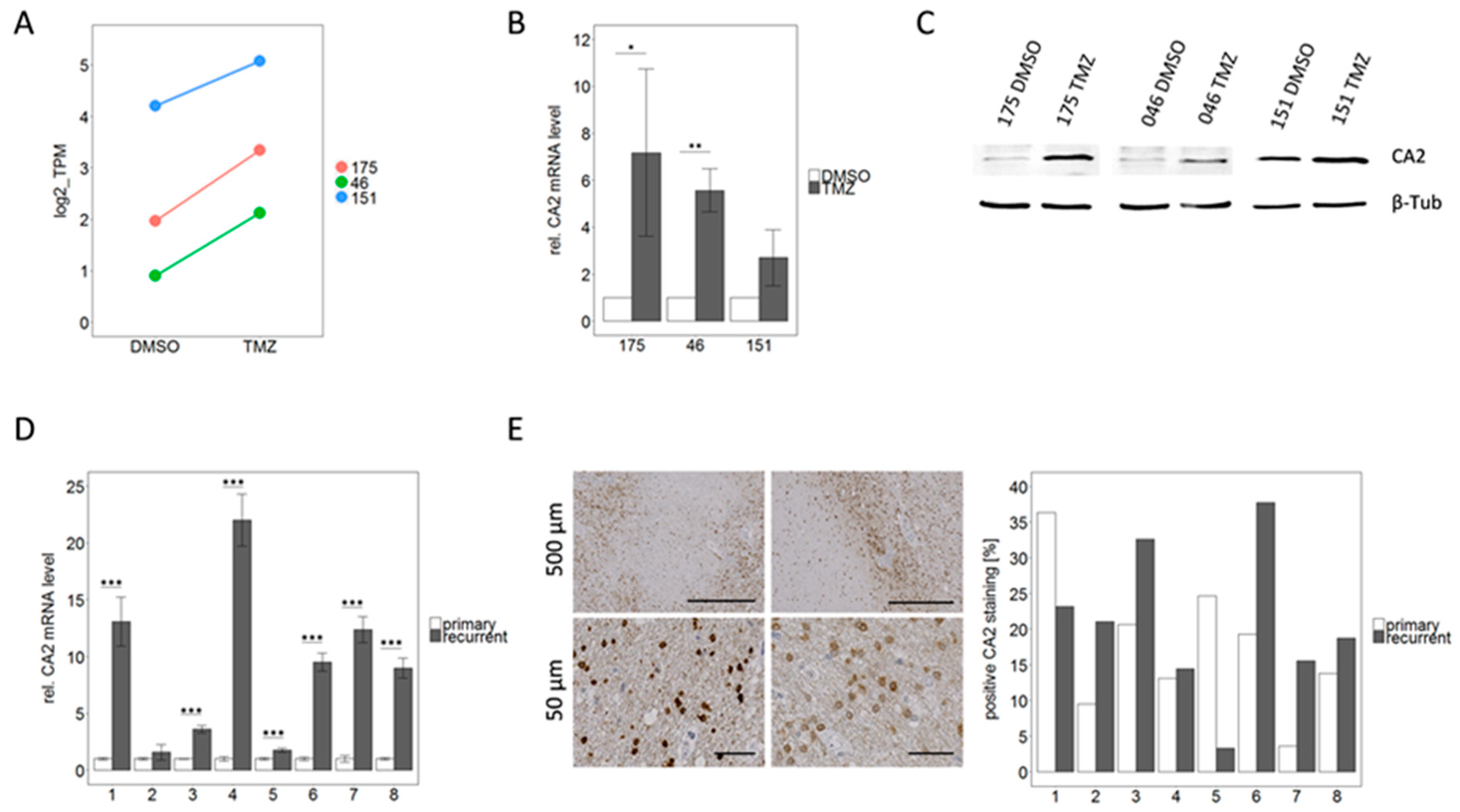
2.4. qPCR Based Validation of Differentially Expressed Genes from RNA Sequencing Points to CA2 as the Most Consistent Mediator of TMZ Resistance
2.5. CA2 Overexpression Is Associated with TMZ Resistance in Primary GBM Stem-Like Cells as well as in Patient Matched Tissue Samples of Primary and Recurrent GBMs
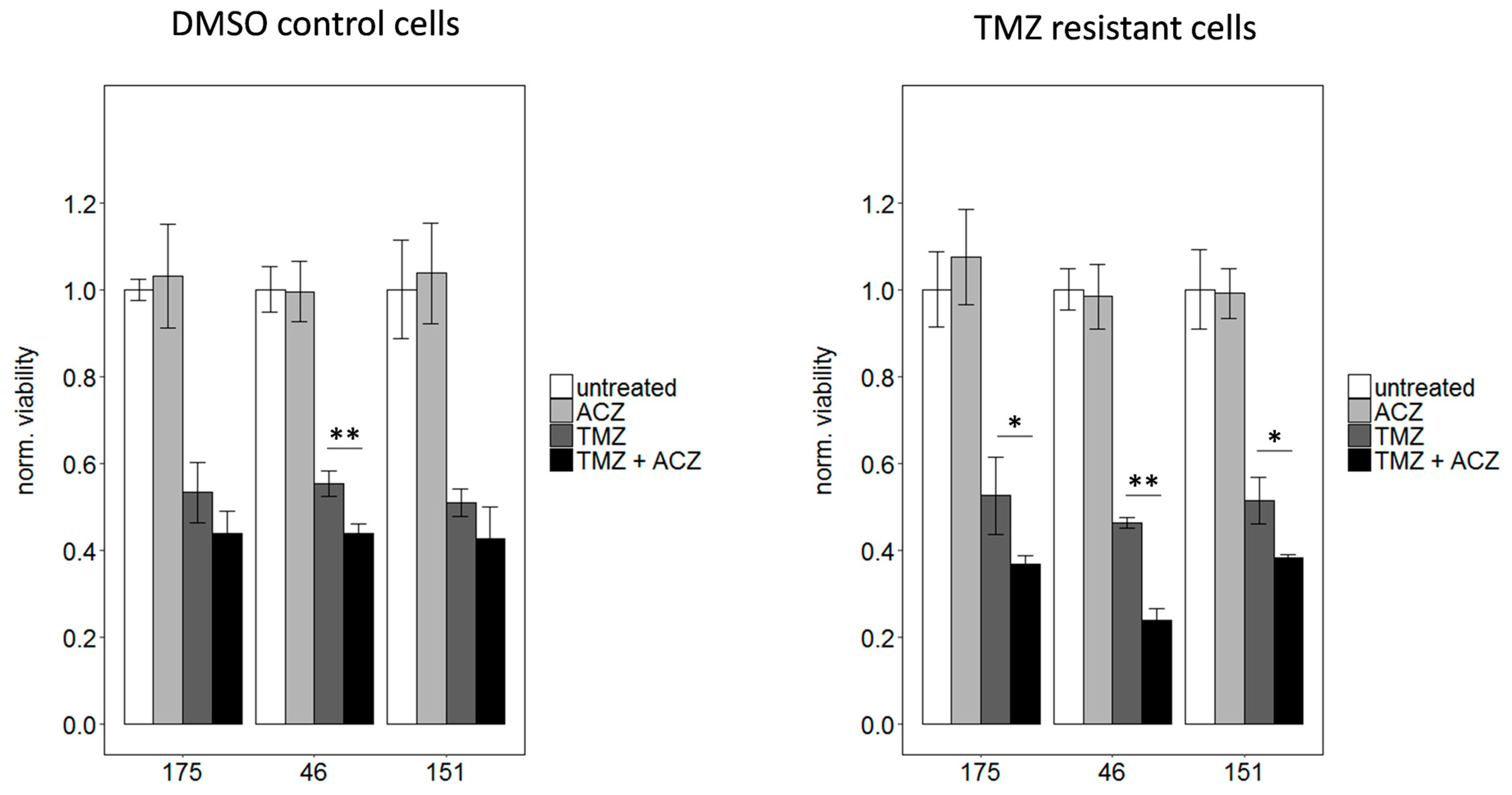
2.6. Inhibition of CA2 Leads to Resensitization of TMZ Resistant Cells
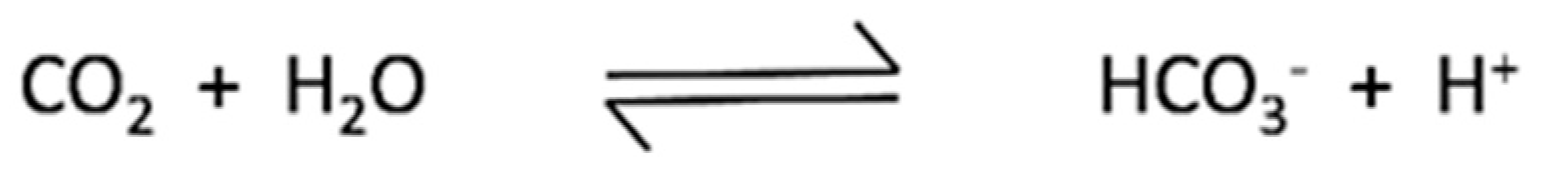
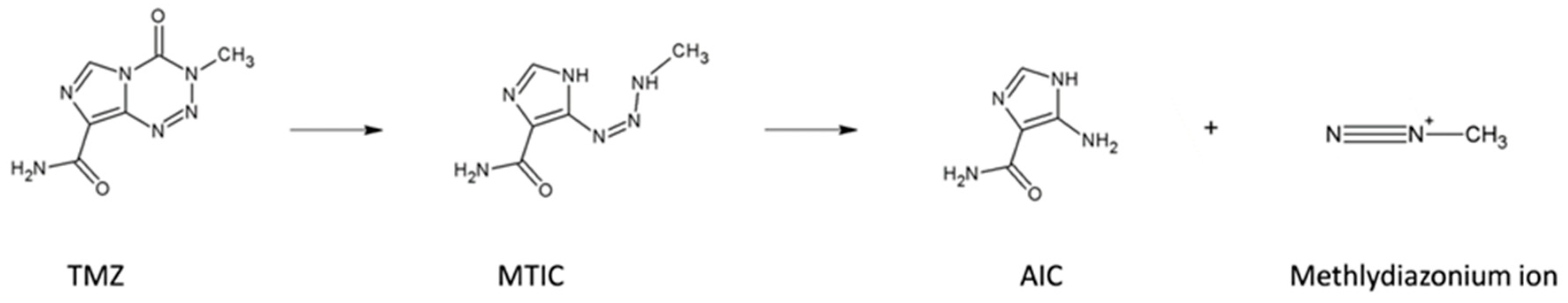
3. Discussion
4. Materials and Methods
4.1. Isolation of Primary GBM Stem-Like Cells
4.2. Cultivation of Primary GBM Stem-Like Cells
4.3. Differentiation Assay
- complete medium + 0.02 ng/µL EGF and bFGF
- complete medium + 1% FCS
- DMEM/F12 base medium + 10% FCS
4.4. Invasion Assay
4.5. Generation of TMZ Resistant Cells
4.6. Viability Assay
4.7. Side Population Analysis
4.8. RNA Extraction and qPCR
4.9. RNA Sequencing, Subsequent Data Analysis, Differential Expression Analysis, and Gene Set Enrichment Analysis (GSEA)
4.10. Protein Extraction and Western Blot
4.11. Immunohistochemistry
4.12. Stereotactic Injection and In Vivo Analysis of Tumor Growth
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HGNC Symbol | Basemean | log2-Fold Change (TMZ vs. DMSO) | lfcSE | Stat | p-Value | padj |
|---|---|---|---|---|---|---|
| TMEM132E | 227.48 | 2.210228 | 0.336322 | 6.571757 | 4.97E-11 | 7.08E-07 |
| ACAP2 | 778.49 | 1.083982 | 0.182214 | 5.948946 | 2.70E-09 | 1.92E-05 |
| C4A | 629.64 | 1.669669 | 0.287942 | 5.798628 | 6.69E-09 | 3.17E-05 |
| DCLRE1C | 171.97 | 1.398863 | 0.260390 | 5.372189 | 7.78E-08 | 0.000243 |
| LY6E | 2315.11 | −0.935013 | 0.174599 | −5.355198 | 8.55E-08 | 0.000243 |
| CNR1 | 2708.31 | 1.336642 | 0.261834 | 5.104927 | 3.31E-07 | 0.000785 |
| DCN | 109.90 | 1.622171 | 0.329603 | 4.921592 | 8.58E-07 | 0.001747 |
| COL2A1 | 2089.18 | −1.554484 | 0.335348 | −4.635431 | 3.56E-06 | 0.006341 |
| WLS | 7274.36 | 0.657914 | 0.143069 | 4.598593 | 4.25E-06 | 0.006731 |
| TRDMT1 | 225.65 | 1.195978 | 0.271745 | 4.401103 | 1.08E-05 | 0.015339 |
| GTPBP1 | 3736.84 | −0.683161 | 0.157797 | −4.329361 | 1.50E-05 | 0.016730 |
| SHISAL1 | 734.77 | −1.170014 | 0.270540 | −4.324744 | 1.53E-05 | 0.016730 |
| AFAP1L2 | 242.41 | −1.241708 | 0.286406 | −4.335486 | 1.45E-05 | 0.016730 |
| ATP1A2 | 5377.06 | −1,331312 | 0.313245 | −4.250073 | 2.14E-05 | 0.021739 |
| CHI3L2 | 119.74 | 1.360969 | 0.330018 | 4.123923 | 3.72E-05 | 0.027242 |
| NKAIN1 | 1452.17 | −0.875030 | 0.211924 | −4.128970 | 3.64E-05 | 0.027242 |
| CA2 | 560.03 | 1.089472 | 0,263365 | 4.136738 | 3.52E-05 | 0.027242 |
| RDH10 | 561.92 | 1.210452 | 0.293958 | 4.117770 | 3.83E-05 | 0.027242 |
| ZNF649 | 597.00 | 0.781817 | 0.188570 | 4,146019 | 3.38E-05 | 0.027242 |
| RTN4R | 1218.69 | −0.946651 | 0.231995 | −4,080488 | 4.49E-05 | 0.028331 |
| NTSR1 | 337.36 | −1.368607 | 0.335840 | −4.075182 | 4.60E-05 | 0.028331 |
| PDGFRA | 1787.36 | 1.343146 | 0.330302 | 4.066417 | 4.77E-05 | 0.028331 |
| ABCA8 | 711.92 | 1.350069 | 0.330592 | 4.083792 | 4.43E-05 | 0.028331 |
| ZNF367 | 799.94 | 0.791751 | 0.195447 | 4.050978 | 5.10E-05 | 0.029056 |
| HAGHL | 379.10 | −1.112411 | 0.276561 | −4.022301 | 5.76E-05 | 0.031569 |
| DNAJB1 | 4917.88 | 0.648952 | 0,164399 | 3.947422 | 7.90E-05 | 0.041670 |
| TSPAN7 | 3715.86 | −0.833058 | 0.212458 | −3.921040 | 8.82E-05 | 0.044846 |
| SHC3 | 2376.20 | 0.768301 | 0.196690 | 3.906150 | 9.38E-05 | 0.046055 |
| TMC6 | 218.99 | −1.006040 | 0.262123 | −3.838046 | 0.000124 | 0.056976 |
| ADAMTS3 | 871.58 | −1.157211 | 0.301136 | −3.842823 | 0.000122 | 0.056976 |
| SCRN2 | 683.63 | −0.800912 | 0.209596 | −3.821212 | 0.000133 | 0.059103 |
| POLR1D | 1643.68 | −0.959674 | 0.252298 | −3.803734 | 0.000143 | 0.061513 |
| MAPT | 651.68 | 1.198264 | 0.315646 | 3.796220 | 0.000147 | 0.061542 |
| PCDH9 | 1621.19 | 0.572030 | 0.151856 | 3.766929 | 0.000165 | 0.063615 |
| DPP4 | 472.82 | 1.143557 | 0.303463 | 3.768355 | 0.000164 | 0.063615 |
| ZNF43 | 748.32 | 0.790207 | 0.209750 | 3.767374 | 0.000165 | 0.063615 |
| BCORL1 | 1470.42 | −0.606643 | 0.162327 | −3.737167 | 0.000186 | 0.069145 |
| F13A1 | 29.81 | −1.003468 | 0.269197 | −3.727634 | 0.000193 | 0.069145 |
| ELFN2 | 176.81 | −1.224097 | 0.329579 | −3.714121 | 0.000204 | 0.069145 |
| CDK5R1 | 1225.39 | −0.668456 | 0.179726 | −3.719317 | 0.000200 | 0.069145 |
| AMBN | 133.27 | −1.090870 | 0.293190 | −3.720695 | 0.000199 | 0.069145 |
| ATG16L2 | 82.97 | −1.188835 | 0.322555 | −3.685686 | 0.000228 | 0.073828 |
| HAS2 | 431.71 | 0.839417 | 0.227706 | 3.686413 | 0.000227 | 0.073828 |
| NBPF1 | 978.13 | 0.724568 | 0.197311 | 3.672207 | 0.000240 | 0.076104 |
| PLK2 | 1290.54 | −0.724093 | 0.198167 | −3.653953 | 0.000258 | 0.079951 |
| PTPRN2 | 27.00 | 0.922263 | 0.254566 | 3.622883 | 0.000291 | 0.088282 |
| WNK4 | 180.70 | 1.208069 | 0.335698 | 3.598675 | 0.000320 | 0.094900 |
| NBPF20 | 181.35 | 1.057601 | 0.295316 | 3.581247 | 0.000342 | 0.097727 |
| MZT2A | 1541.96 | −0.801742 | 0.223927 | −3.580381 | 0.000343 | 0.097727 |
References
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Eng. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Ulasov, I.V.; Lesniak, M.S. Virotherapy against malignant glioma stem cells. Cancer Lett. 2010, 289, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, F.H.; Pruitt, A. Assumptions in the radiotherapy of glioblastoma. Neurology 1980, 30, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Akbari-Birgani, S.; Paranjothy, T.; Zuse, A.; Janikowski, T.; Cieślar-Pobuda, A.; Likus, W.; Urasińska, E.; Schweizer, F.; Ghavami, S.; Klonisch, T.; et al. Cancer stem cells, cancer-initiating cells and methods for their detection. Drug Discov. Today 2016, 21, 836–842. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Mollapour Sisakht, M.; Azizi, Z.; Taghikhani, A.; Łos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updat. 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Ahmad, G.; Amiji, M.M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin. Drug. Deliv. 2017, 14, 997–1008. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41:1–41:7. [Google Scholar] [CrossRef] [PubMed]
- Van Thuijl, H.F.; Mazor, T.; Johnson, B.E.; Fouse, S.D.; Aihara, K.; Hong, C.; Malmström, A.; Hallbeck, M.; Heimans, J.J.; Kloezeman, J.J.; et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta. Neuropathol. 2015, 129, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Nordfors, K.; Haapasalo, J.; Korja, M.; Niemelä, A.; Laine, J.; Parkkila, A.K.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. The tumour-associated carbonic anhydrases CA II, CA IX and CA XII in a group of medulloblastomas and supratentorial primitive neuroectodermal tumours: an association of CA IX with poor prognosis. BMC Cancer 2010, 10, 148:1–148:10. [Google Scholar] [CrossRef] [PubMed]
- Nortunen, M.; Huhta, H.; Helminen, O.; Parkkila, S.; Kauppila, J.H.; Karttunen, T.J.; Saarnio, J. Carbonic anhydrases II, IX, and XII in Barrett’s esophagus and adenocarcinoma. Virchows Arch. 2018, 473, 567–575. [Google Scholar] [CrossRef]
- Karjalainen, S.L.; Haapasalo, H.K.; Aspatwar, A.; Barker, H.; Parkkila, S.; Haapasalo, J.A. Carbonic anhydrase related protein expression in astrocytomas and oligodendroglial tumors. BMC Cancer 2018, 18, 584:1–584:9. [Google Scholar] [CrossRef]
- Haapasalo, J.; Nordfors, K.; Järvelä, S.; Bragge, H.; Rantala, I.; Parkkila, A.K.; Haapasalo, H.; Parkkila, S. Carbonic anhydrase II in the endothelium of glial tumors: a potential target for therapy. Neuro. Oncol. 2007, 9, 308–313. [Google Scholar] [CrossRef]
- Supuran, C.T.; Briganti, F.; Tilli, S.; Chegwidden, W.R.; Scozzafava, A. Carbonic Anhydrase Inhibitors: Sulfonamides as Antitumor Agents? Bioorg. Med. Chem. 2001, 9, 703–714. [Google Scholar] [CrossRef]
- Das, A.; Banik, N.L.; Ray, S.K. Modulatory effects of acetazolomide and dexamethasone on temozolomide-mediated apoptosis in human glioblastoma T98G and U87MG cells. Cancer Invest. 2008, 26, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Amiri, A.; Le, P.U.; Moquin, A.; Machkalyan, G.; Petrecca, K.; Gillard, J.W.; Yoganathan, N.; Maysinger, D. Inhibition of carbonic anhydrase IX in glioblastoma multiforme. Eur. J. Pharm. Biopharm. 2016, 109, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Bernal, G.M.; Cahill, K.E.; Pytel, P.; Fitzpatrick, C.A.; Mashek, H.; Weichselbaum, R.R.; Yamini, B. BCL3 expression promotes resistance to alkylating chemotherapy in gliomas. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- A phase I Study of Safety and Tolerability of Acetazolamide with Temozolomide in Adults with newly diagnosed MGMT Promotor Methylated Malignant Glioma (NCT03011671). Available online: https://clinicaltrials.gov/ct2/show/NCT03011671 (accessed on 3 April 2019).
- Swietach, P. What is pH regulation, and why do cancer cells need it? Cancer Metastasis Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Stéphanou, A.; Ballesta, A. pH as a potential therapeutic target to improve temozolomide antitumor efficacy: A mechanistic modeling study. Pharmacol. Res. Perspect. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Liu, S.D.; Liu, J.T.; Holden, S.A.; Herman, T.S. A carbonic anhydrase inhibitor as a potential modulator of cancer therapies. Anticancer Res. 1993, 13, 1549–1556. [Google Scholar] [PubMed]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro. Oncol. 2012, 14, 1358–1366. [Google Scholar] [CrossRef]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Hannen, R.; Steffani, M.; Voellger, B.; Carl, B.; Wang, J.; Bartsch, J.W.; Nimsky, C. Effects of anti-estrogens on cell invasion and survival in pituitary adenoma cells: A systematic study. J. Steroid Biochem. Mol. Biol. 2019, 187, 88–96. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521:1–1521:19. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550:1–550:21. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016. [Google Scholar] [CrossRef]
- Powers, R.K.; Goodspeed, A.; Pielke-Lombardo, H.; Tan, A.C.; Costello, J.C. GSEA-InContext: identifying novel and common patterns in expression experiments. Bioinformatics 2018, 34, i555–i564. [Google Scholar] [CrossRef] [PubMed]
- Gene Set Enrichment Analysis (GSEA)–Molecular Signature Database (MSigDB). Available online: http://software.broadinstitute.org/gsea/msigdb/index.jsp (accessed on 3 April 2019).
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017, 7, 16878:1–16878:7. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]








| No. | Sex | Age | MGMT Status | IDH1 R132H |
|---|---|---|---|---|
| 175 | m | 65 | methylated | wildtype |
| 46 | f | 43 | methylated | wildtype |
| 151 | m | 66 | methylated | wildtype |
| Concentration (µM) | Amount DMSO (%) |
|---|---|
| 5 | 0.0097 |
| 10 | 0.019 |
| 15 | 0.029 |
| 20 | 0.039 |
| 50 | 0.097 |
| 100 | 0.19 |
| 200 | 0.39 |
| 300 | 0.58 |
| 400 | 0.78 |
| 500 | 0.97 |
| 750 | 1.46 |
| 1000 | 1.94 |
| Pair | Sex | Age | Survival | Latency | MGMT Status | EGFR v III | IDH1 R132H | Ki67 |
|---|---|---|---|---|---|---|---|---|
| 1 | m | 51 | 371 | 172 | not methylated | positive | negative | 25% |
| not methylated | negative | negative | 10% | |||||
| 2 | m | 52 | 760 | 491 | not methylated | positive | negative | 75% |
| not methylated | negative | negative | 20% | |||||
| 3 | f | 51 | 445 | 147 | not methylated | negative | negative | 20% |
| not methylated | negative | negative | 5% | |||||
| 4 | m | 57 | 343 | 288 | not methylated | negative | negative | up to 50% |
| not methylated | negative | negative | 10% | |||||
| 5 | f | 77 | 550 | 266 | not methylated | negative | negative | 15% |
| not methylated | positive | negative | 7% | |||||
| 6 | m | 48 | 524 | 603 | not methylated | negative | negative | 25% |
| not methylated | negative | negative | up to 50% | |||||
| 7 | m | 71 | 532 | 357 | Methylated | positive | negative | 50% |
| not methylated | negative | negative | 50% | |||||
| 8 | m | 45 | 576 | 397 | not methylated | negative | negative | 30% |
| not methylated | negative | negative | up to 5% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 2019, 11, 921. https://doi.org/10.3390/cancers11070921
Hannen R, Selmansberger M, Hauswald M, Pagenstecher A, Nist A, Stiewe T, Acker T, Carl B, Nimsky C, Bartsch JW. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers. 2019; 11(7):921. https://doi.org/10.3390/cancers11070921
Chicago/Turabian StyleHannen, Ricarda, Martin Selmansberger, Maria Hauswald, Axel Pagenstecher, Andrea Nist, Thorsten Stiewe, Till Acker, Barbara Carl, Christopher Nimsky, and Jörg Walter Bartsch. 2019. "Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor" Cancers 11, no. 7: 921. https://doi.org/10.3390/cancers11070921
APA StyleHannen, R., Selmansberger, M., Hauswald, M., Pagenstecher, A., Nist, A., Stiewe, T., Acker, T., Carl, B., Nimsky, C., & Bartsch, J. W. (2019). Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers, 11(7), 921. https://doi.org/10.3390/cancers11070921