Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
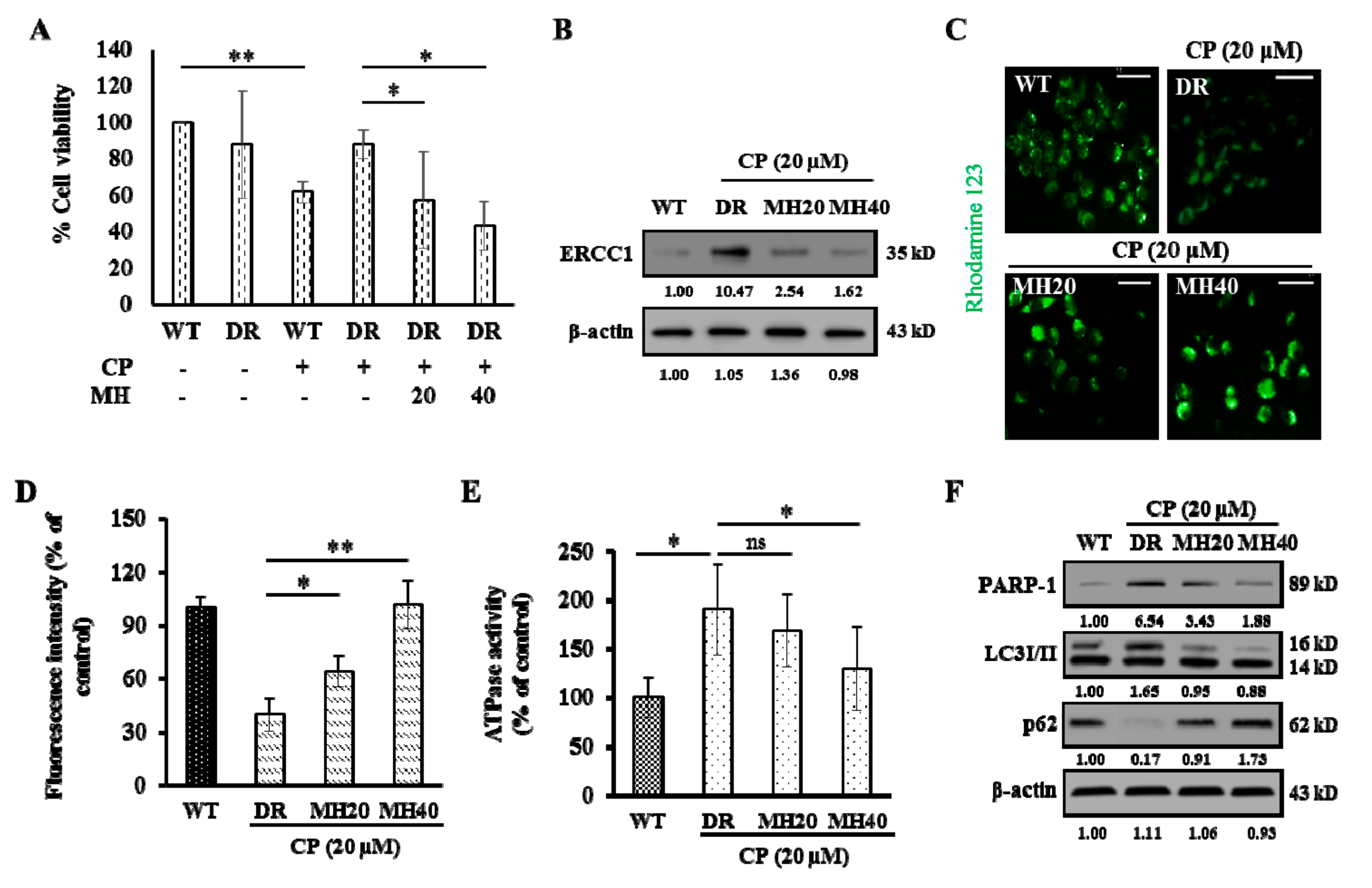
2.1. MH Treatment Reverses an Acquired CP-Resistant Phenotype of HepG2DR Cells
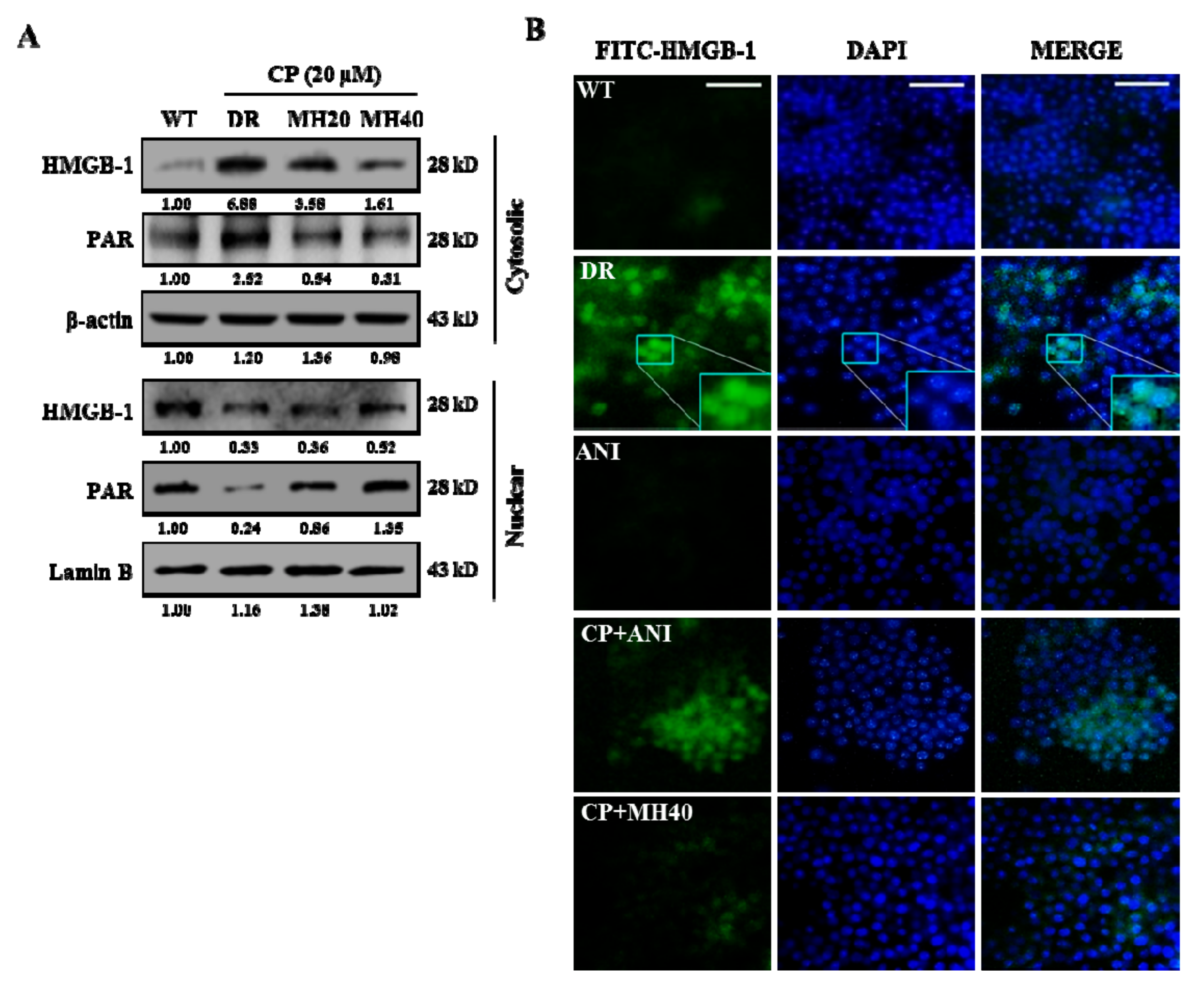
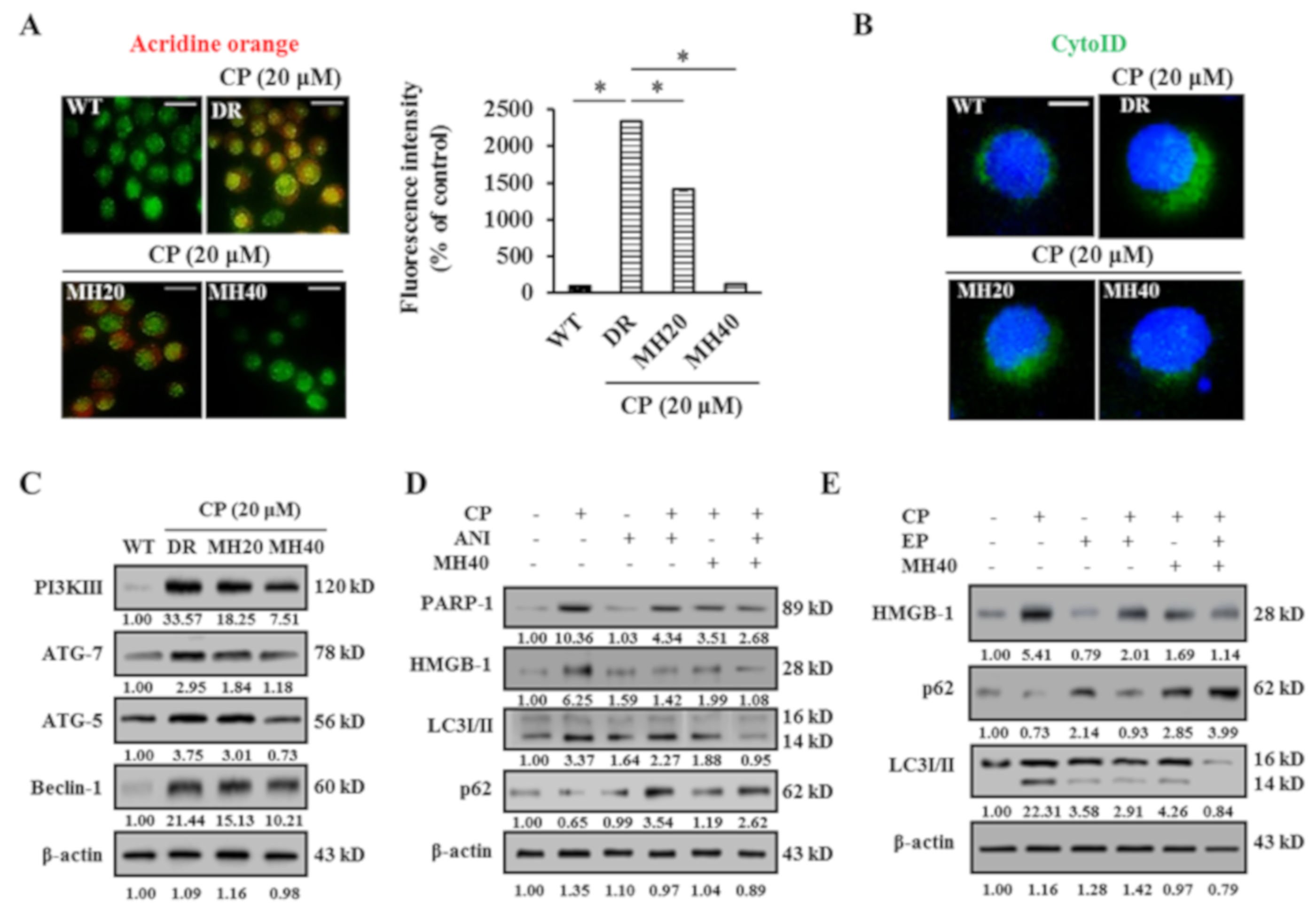
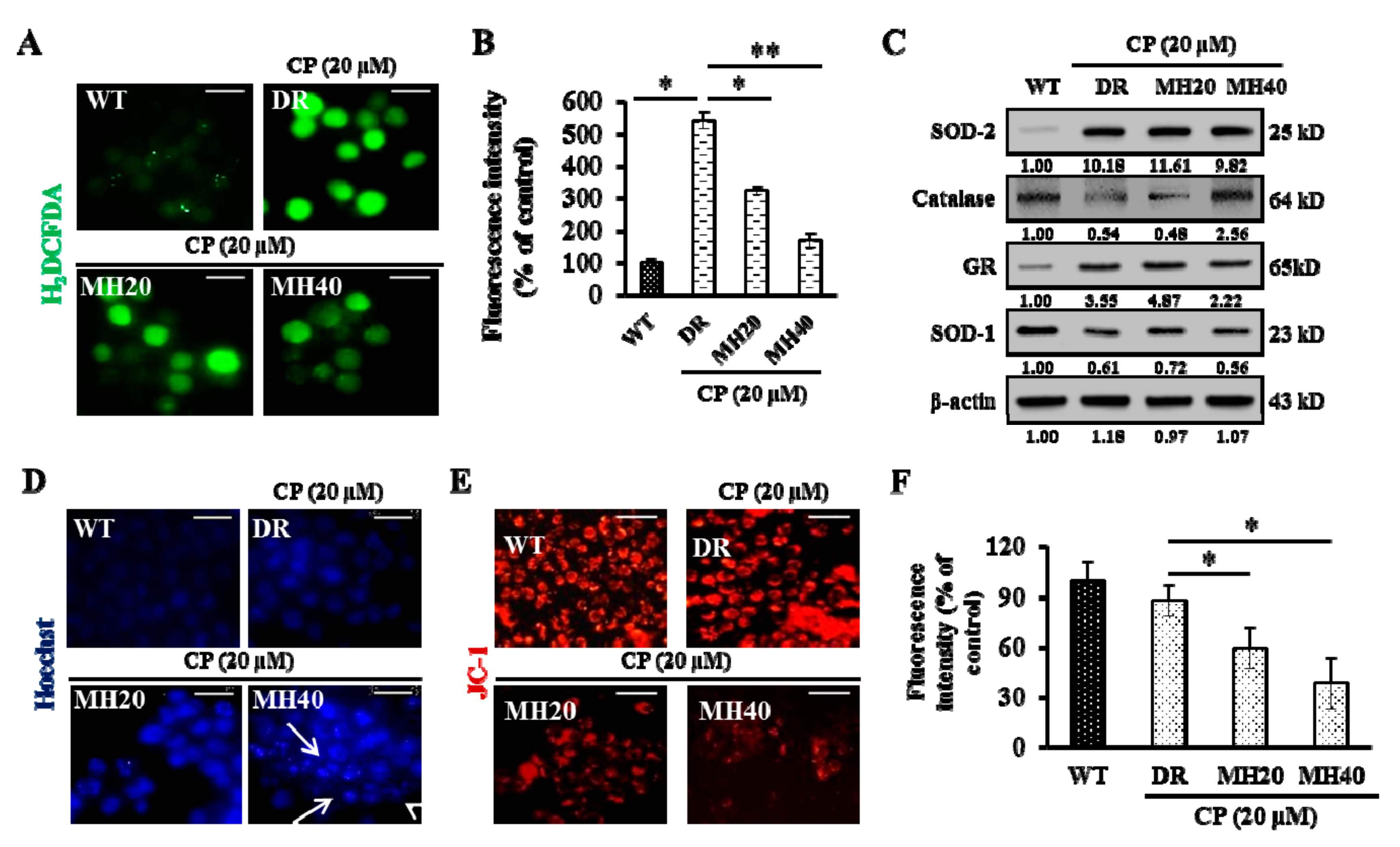
2.2. MH Suppresses PARP1-Mediated Autophagy Activation in HepG2DR Cells
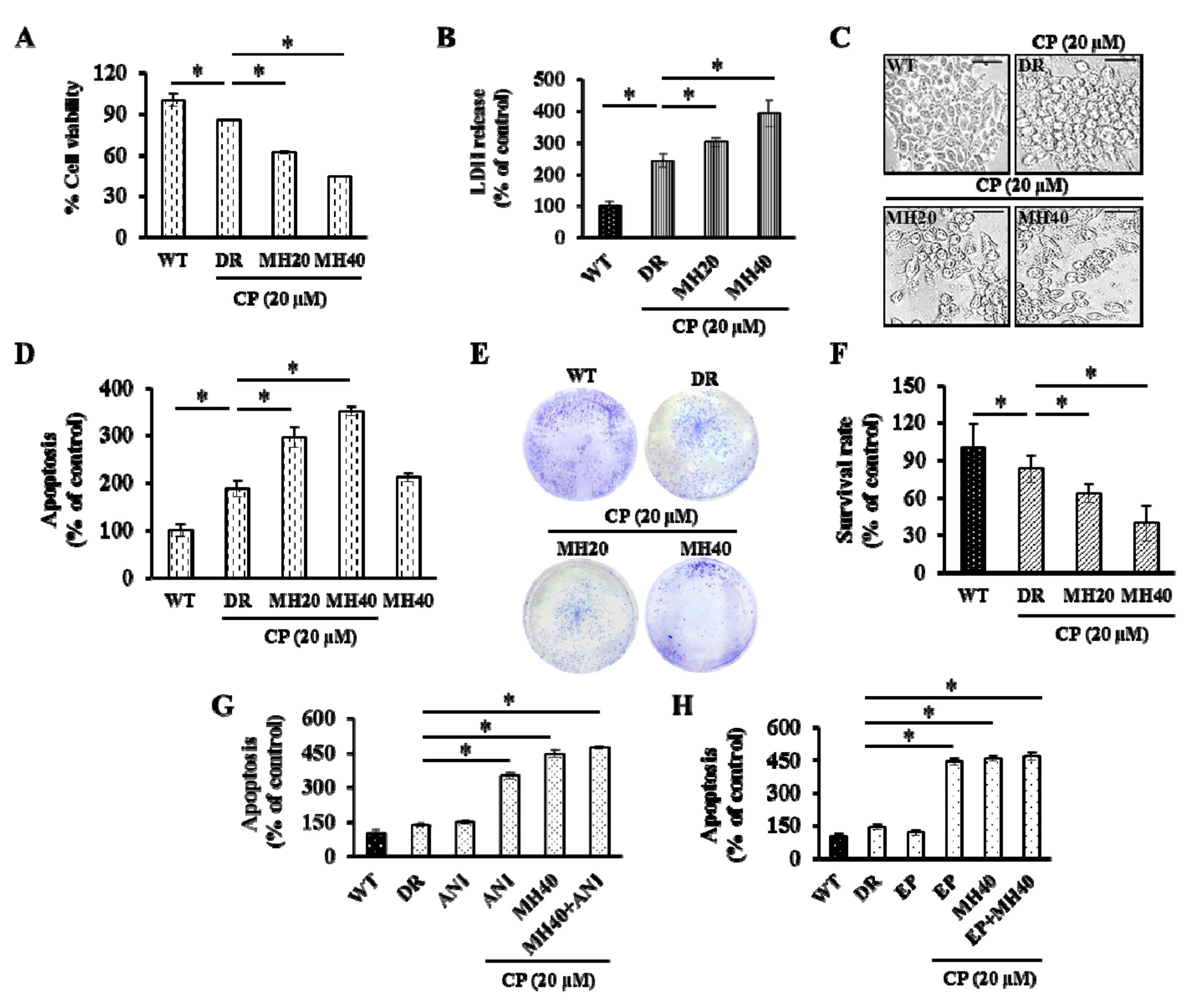
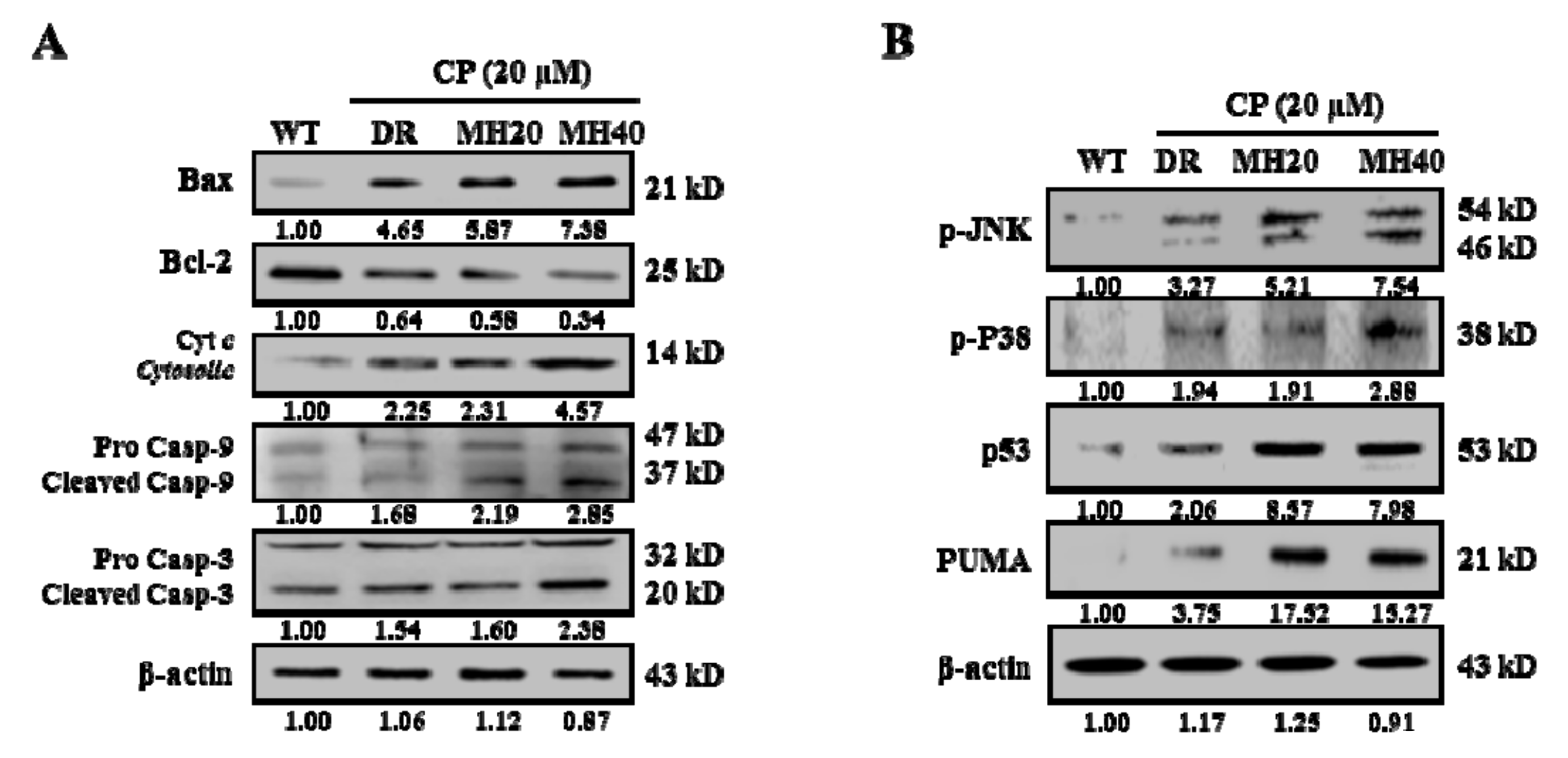
2.3. MH Enhances Apoptosis of HepG2DR Cells against CP by Suppressing PARP1-Mediated Autophagy
2.4. MH Promotes the Mitochondria-Mediated Intrinsic Pathway of CP-Induced Apoptosis in HepG2DR Cells
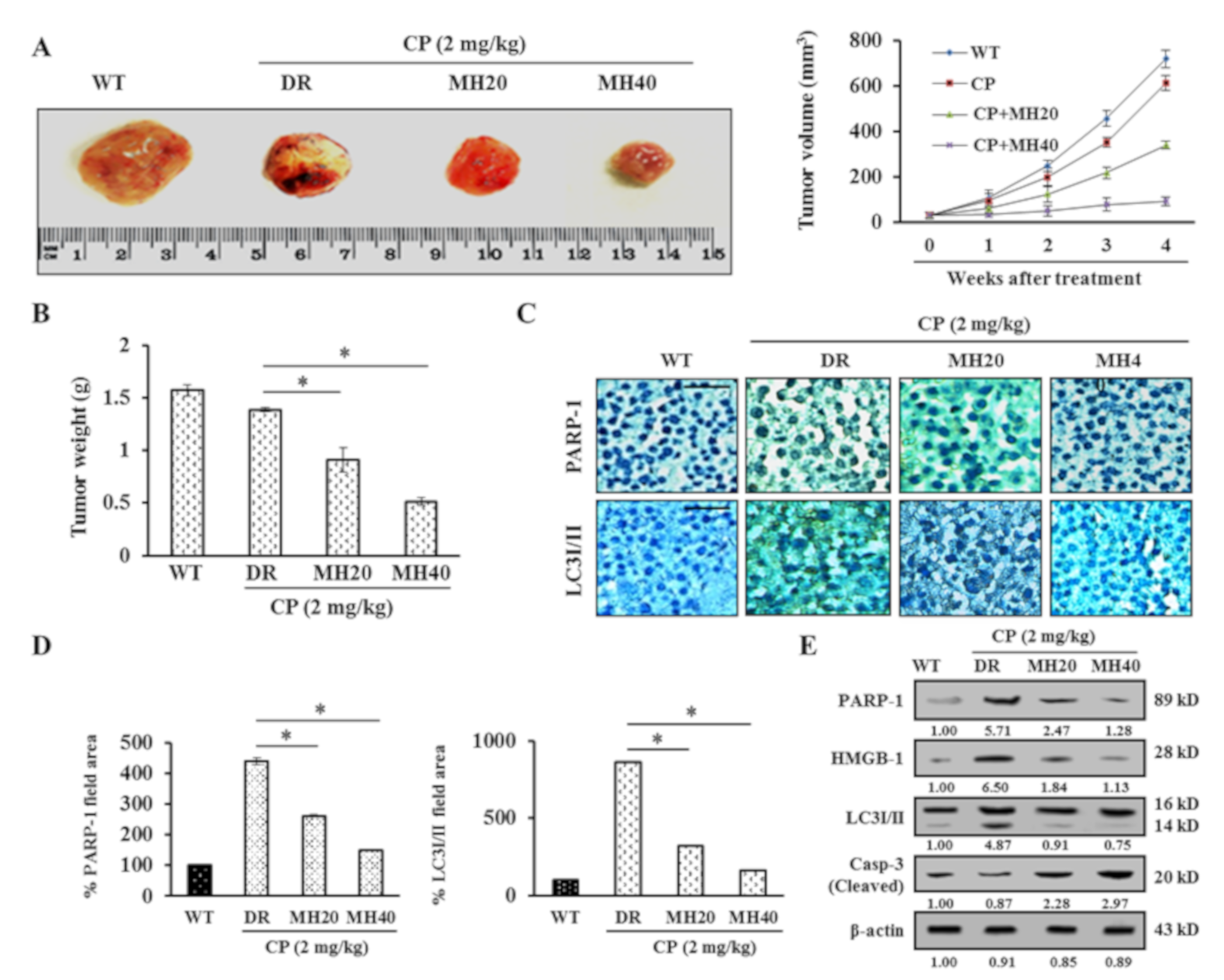
2.5. MH-Mediated PARP1 Regulation Potentiates the Antitumor Effect of CP in HepG2DR Hepatoma-Bearing Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. ATPase Activity Assay
4.4. Cell Viability Assay
4.5. Apoptosis Assay
4.6. Colony Formation Assay
4.7. Determination of Autophagy
4.8. Determination of Intracellular ROS
4.9. Isolation of Mitochondrial Proteins
4.10. Determination of Mitochondrial Membrane Potential
4.11. Immunoblot Analysis
4.12. Detection of Nuclear DNA Condensation with Hoechst-33342
4.13. Quantitative Real-Time PCR Analysis
4.14. Immunostaining
4.15. siRNA Transfection
4.16. Animals and Establishment of a Mouse Xenograft Model
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, Y.; Zhang, Y.; Ge, L.; Kwok, H.F. The roles of protein tyrosine phosphatases in Hepatocellular carcinoma. Cancers 2018, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Treatment of advanced hepatocellular carcinoma with emphasis on hepatic arterial infusion chemotherapy and molecular targeted therapy. Liver Cancer 2012, 1, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Muñoz-Gámez, J.A.; Rodríguez-Vargas, J.M.; Quiles-Pérez, R.; Aguilar-Quesada, R.; Martín-Oliva, D. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef]
- Borst, P.; Rottenberg, S.; Jonkers, J. How do real tumors become resistant to cisplatin? Cell Cycle 2008, 7, 1353–1359. [Google Scholar] [CrossRef]
- Florea, A.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Henkels, K.M.; Turchi, J.J. Induction of apoptosis in cisplatin-sensitive and -resistant human ovarian cancer cell lines. Cancer Res. 1997, 57, 4488–4492. [Google Scholar] [PubMed]
- Johnson, S.W.; Shen, D.; Pastan, I.; Gottesman, M.M.; Hamilton, T.C. Cross-resistance, cisplatin accumulation, and platinum-DNA adduct formation and removal in cisplatin-sensitive and -resistant human hepatoma cell lines. Exp. Cell Res. 1996, 226, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Dabholkar, M.; Vionnet, J.; Bostick-Bruton, F.; Yu, J.J.; Reed, E. Messenger RNA levels of XPAC and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J. Clin. Investig. 1994, 94, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Liu, L.; Dai, W.; Zhang, W.; Yang, Y.; Wang, H. Xeroderma Pigmentosum Group A Promotes Autophagy to Facilitate Cisplatin Resistance in Melanoma Cells through the Activation of PARP1. J. Investig. Dermatol. 2016, 136, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Elmansi, A.M.; El-Karef, A.A.; Shishtawy, M.M.E.; Eissa, L.A. Hepatoprotective Effect of Curcumin on Hepatocellular Carcinoma through Autophagic and Apoptic Pathways. Ann. Hepatol. 2017, 16, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Shen, C.; Li, C.; Liu, Y.; Gao, D.; Shi, C. Inhibition of autophagy induced by quercetin at a late stage enhances cytotoxic effects on glioma cells. Tumor Biol. 2016, 37, 3549–3560. [Google Scholar] [CrossRef]
- Caselli, A.; Cirri, P.; Santi, A.; Paoli, P. Morin: A Promising Natural Drug. Curr. Med. Chem. 2016, 23, 774–791. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Simic, M.G. Antioxidants in nutrition. Ann. N. Y. Acad. Sci. 2000, 899, 326–334. [Google Scholar] [CrossRef]
- Jakhar, R.; Paul, S.; Chauhan, A.K.; Kang, S.C. Morin hydrate augments phagocytosis mechanism and inhibits LPS induced autophagic signaling in murine macrophage. Int. Immunopharmacol. 2014, 22, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Chauhan, A.K.; Kang, S.C. Morin hydrate ameliorates cisplatin-induced ER stress, inflammation and autophagy in HEK-293 cells and mice kidney via PARP-1 regulation. Int. Immunopharmacol. 2018, 56, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Jakhar, R.; Kang, S.C. Morin hydrate attenuates the acrylamide-induced imbalance in antioxidant enzymes in a murine model. Int. J. Mol. Med. 2015, 36, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Chu, A.C.; Fung, K.P. Inhibition of P-glycoprotein expression and reversal of drug resistance of human hepatoma HepG2 cells by multidrug resistance gene (mdr1) antisense RNA. Life Sci. 2000, 67, 2117–2224. [Google Scholar] [CrossRef]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; André, F.; Haddad, V. IALT Bio Investigators, DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Cho, H.J.; Paul, S.; Jakhar, R.; Khan, I.; Lee, S.J. Vitexin induces apoptosis by suppressing autophagy in multi-drug resistant colorectal cancer cells. Oncotarget 2017, 9, 3278–3291. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, J.M.; Chwae, Y.J.; Kim, Y.H.; Lee, J.H.; Kim, K.; Lee, T.H.; Kim, S.J.; Park, J.H. Cisplatin-induced apoptosis in Hep3B cells: Mitochondria-dependent and -independent pathways. Biochem. Pharmacol. 2004, 67, 1459–1468. [Google Scholar] [CrossRef]
- Ishikawa, T. Future perspectives on the treatment of hepatocellular carcinoma with cisplatin. World J. Hepatol. 2009, 1, 8–16. [Google Scholar] [CrossRef]
- Rosland, G.V.; Engelsen, A.S. Novel points of attack for targeted cancer therapy. Basic Clin. Pharmacol. Toxicol. 2014, 116, 9–18. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Divac Rankov, A.; Ljujić, M.; Petrić, M.; Radojković, D.; Pešić, M.; Dinić, J. Targeting autophagy to modulate cell survival: A comparative analysis in cancer, normal and embryonic cells. Histochem. Cell Biol. 2017, 148, 529–544. [Google Scholar] [CrossRef]
- Guo, W.J.; Zhang, Y.M.; Zhang, L.; Huang, B.; Tao, F.F.; Chen, W. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013, 9, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, L.; Xie, M.; Sun, X.; Yu, Y.; Kang, R. Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 2015, 11, 214–224. [Google Scholar] [CrossRef]
- Yang, L.; Yu, Y.; Kang, R.; Yang, M.; Xie, M.; Wang, Z. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk. Lymphoma 2012, 53, 315–322. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, Y.; Ren, X.; Zhang, L.; Yap, K.L.; Wu, H. NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene 2012, 31, 1055–1064. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef]
- Wan, B.; Dai, L.; Wang, L.; Zhang, Y.; Huang, H.; Qian, G.; Yu, T. Knockdown of BRCA2 enhances cisplatin and cisplatin-induced autophagy in ovarian cancer cells. Endocr. Relat. Cancer 2018, 25, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Y.; Liu, Q.; Wang, J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 2011, 28, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Onodera, H.; Ueda, M.; Imai, T.; Hirose, M. A 13-week subchronic toxicity study of dietary administered morin in F344 rats. Food Chem. Toxicol. 2006, 44, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Hwang, Y.S.; Yoon, J.; Lee, M.; Lee, H.G.; Daar, I.O. EphrinB1 promotes cancer cell migration and invasion through the interaction with RhoGDI1. Oncogene 2018, 37, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, J.T.; Lee, S.J.; Hwang, Y.S.; Park, S.Y.; Kim, B.Y. Protein phosphatase 1B dephosphorylates Rho guanine nucleotide dissociation inhibitor 1 and suppresses cancer cell migration and invasion. Cancer Lett. 2018, 417, 141–151. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.P.; Cho, H.J.; Kim, J.-T.; Baek, K.E.; Lee, H.G.; Kang, S.C. Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers 2019, 11, 986. https://doi.org/10.3390/cancers11070986
Singh MP, Cho HJ, Kim J-T, Baek KE, Lee HG, Kang SC. Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers. 2019; 11(7):986. https://doi.org/10.3390/cancers11070986
Chicago/Turabian StyleSingh, Mahendra Pal, Hee Jun Cho, Jong-Tae Kim, Kyoung Eun Baek, Hee Gu Lee, and Sun Chul Kang. 2019. "Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma" Cancers 11, no. 7: 986. https://doi.org/10.3390/cancers11070986
APA StyleSingh, M. P., Cho, H. J., Kim, J.-T., Baek, K. E., Lee, H. G., & Kang, S. C. (2019). Morin Hydrate Reverses Cisplatin Resistance by Impairing PARP1/HMGB1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers, 11(7), 986. https://doi.org/10.3390/cancers11070986