Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
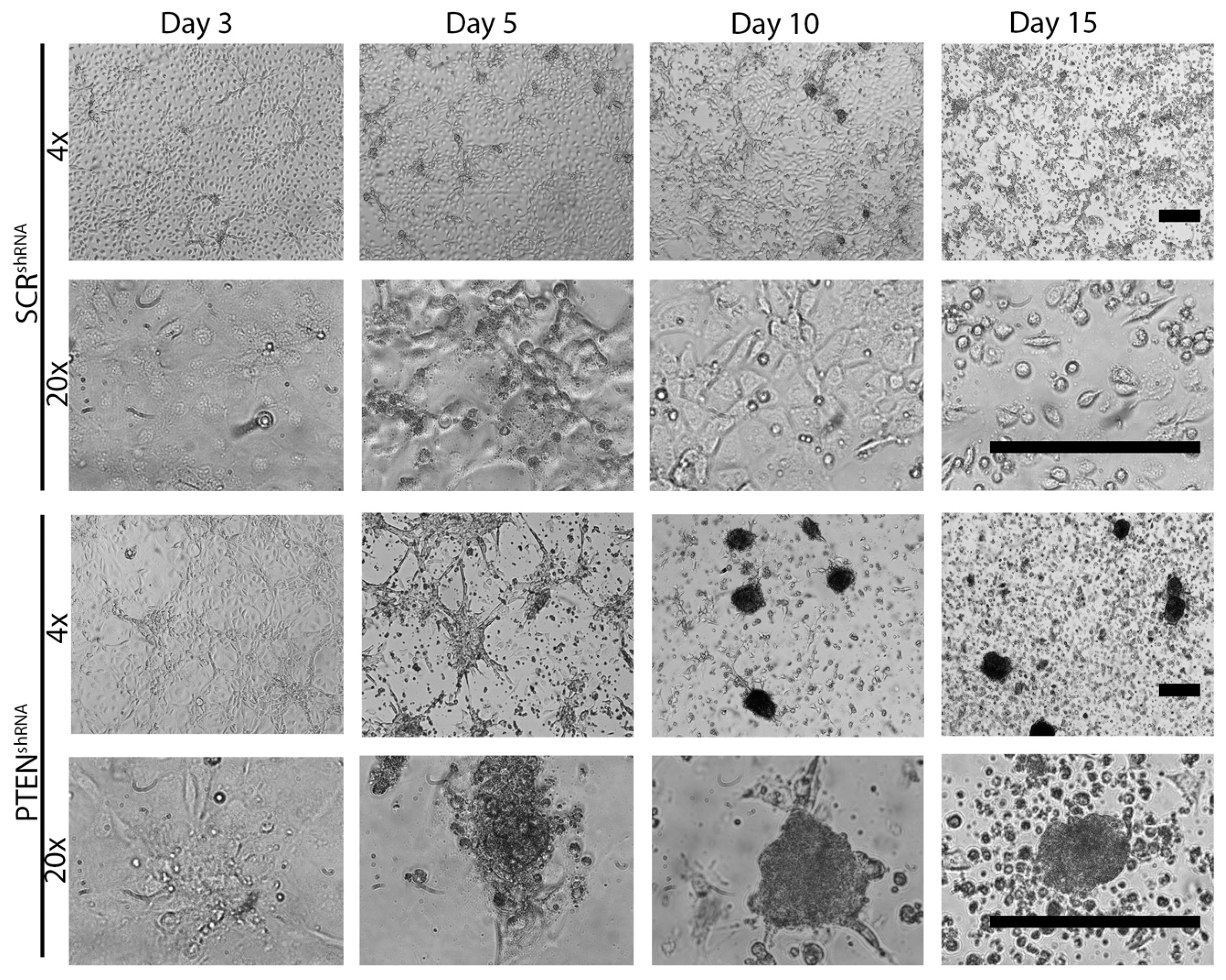
2.1. Loss of PTEN Induces MTS Formation in Vitro and in Vivo
2.2. Wild Type Cells Can Be Incorporated into MTS in Small Numbers
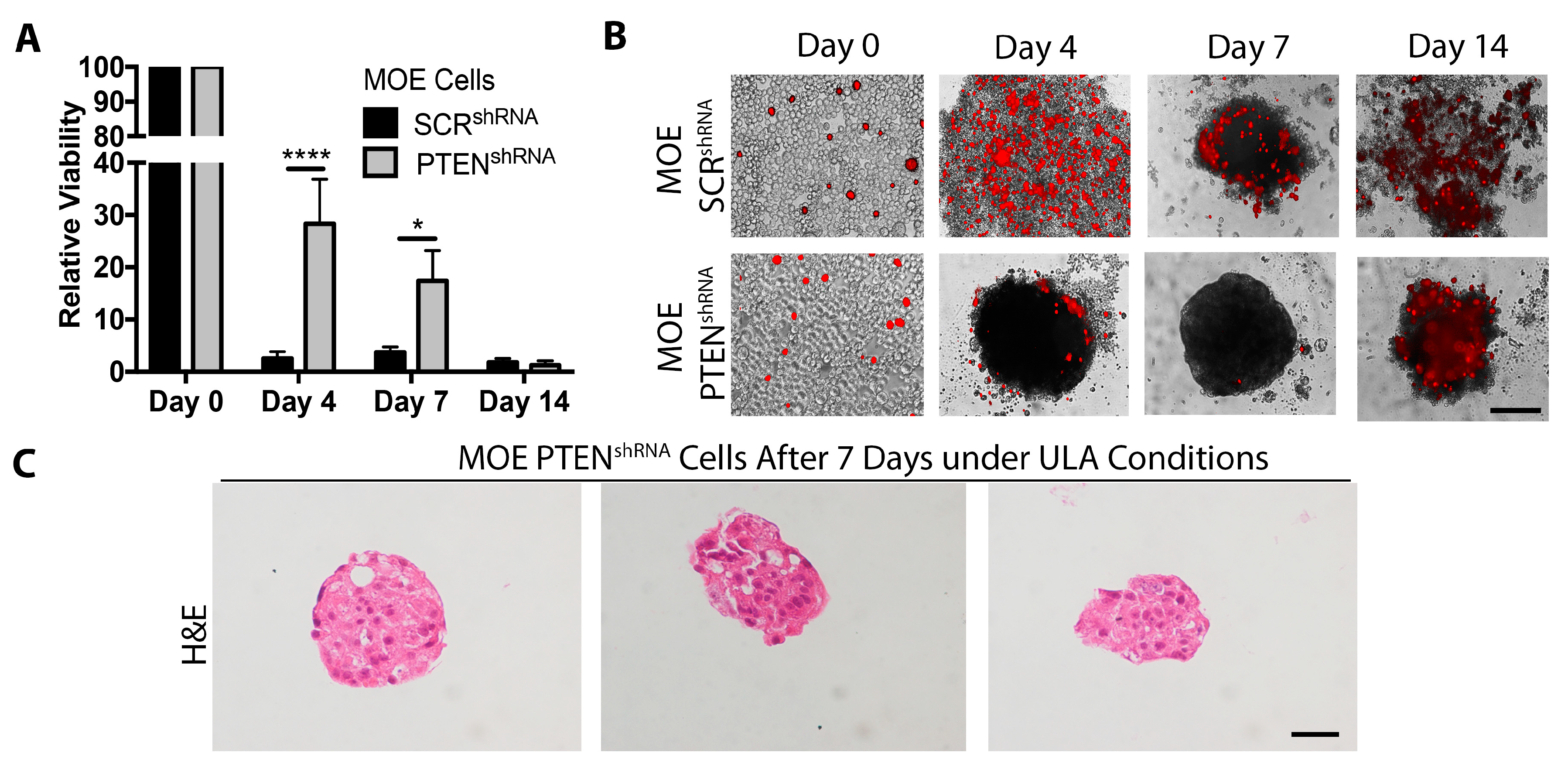
2.3. Loss of PTEN Increases Cell Survival under ULA Conditions
2.4. MTS Attached to Ovarian Stroma Exposed during Ovulation
2.5. MTS Form Tumors after Intrabursal Xenograft
3. Discussion
4. Material and Methods
4.1. Cell Lines and Cell Culture
4.2. MTS Formation
4.3. Viability Assay
4.4. MOSE and MOFIB Attachment Assay
4.5. MTS Clearance Assay
4.6. Hematoxylin and Eosin Staining of MTS
4.7. Mouse Studies
4.8. Ex Vivo Ovarian Attachment Assay
4.9. Transgenic Mice and Immunohistochemistry
4.10. Xenograft Study
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Piek, J.M.J.; Verheijen, R.H.M.; Kenemans, P.; Massuger, L.F.; Bulten, H.; van Diest, P.J. BRCA1/2-related ovarian cancers are of tubal origin: A hypothesis. Gynecol. Oncol. 2003, 90, 491. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Baggerly, K.A.; Patterson, A.P.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin. Cancer Res. 2005, 11, 6116–6126. [Google Scholar] [CrossRef] [PubMed]
- Falconer, H.; Yin, L.; Grönberg, H.; Altman, D. Ovarian cancer risk after salpingectomy: A nationwide population-based study. J. Natl. Cancer Inst. 2015, 107, dju410. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.; Baandrup, L.; Dehlendorff, C.; Kjaer, S.K. Tubal ligation and salpingectomy and the risk of epithelial ovarian cancer and borderline ovarian tumors: A nationwide case-control study. Acta Obstet. Gynecol. Scand. 2015, 94, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Eddie, S.L.; Quartuccio, S.M.; Ó hAinmhire, E.; Moyle-Heyrman, G.; Lantvit, D.D.; Wei, J.-J.; Vanderhyden, B.C.; Burdette, J.E. Tumorigenesis and peritoneal colonization from fallopian tube epithelium. Oncotarget 2015, 21, 20500–20512. [Google Scholar] [CrossRef]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca; Tp53; Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef]
- Russo, A.; Czarnecki, A.; Dean, M.; Modi, D.; Lantvit, D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef]
- Perets, R.; Drapkin, R. It’s totally tubular…riding the new wave of ovarian cancer research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef]
- Dean, M.; Davis, D.A.; Burdette, J.E. Activin A stimulates migration of the fallopian tube epithelium, an origin of high-grade serous ovarian cancer, through non-canonical signaling. Cancer Lett. 2017, 391, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Yang-Hartwich, Y.; Gurrea-Soteras, M.; Sumi, N.; Joo, W.D.; Holmberg, J.C.; Craveiro, V.; Alvero, A.B.; Mor, G. Ovulation and extra-ovarian origin of ovarian cancer. Sci. Rep. 2014, 4, 6116. [Google Scholar] [CrossRef] [PubMed]
- Zink, K.E.; Dean, M.; Burdette, J.E.; Sanchez, L.M. Imaging mass spectrometry reveals crosstalk between the fallopian tube and the ovary that drives primary metastasis of ovarian cancer. ACS Cent. Sci. 2018, 4, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Jin, V.; Russo, A.; Lantvit, D.D.; Burdette, J.E. Exposure of the extracellular matrix and colonization of the ovary in metastasis of fallopian tube derived cancer. Carcinogenesis 2019, 40, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Edwards, N.S.; Yates, J.D. Spheroids and cell survival. Crit. Rev. Oncol. Hematol. 2000, 36, 61–74. [Google Scholar] [CrossRef]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P.N. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Casey, R.C.; Burleson, K.M.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Ruff, L.E.; Skubitz, A.P.N. β1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am. J. Pathol. 2001, 159, 2071–2080. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Habyan, S.A.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef]
- Bijron, J.G.; Seldenrijk, C.A.; Zweemer, R.P.; Lange, J.G.; Verheijen, R.H.M.; van Diest, P.J. Fallopian tube intraluminal tumor spread from noninvasive precursor lesions: A novel metastatic route in early pelvic carcinogenesis. Am. J. Surg. Pathol. 2013, 37, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of ovarian cancer progression reveals diverse metastatic trajectories including intraepithelial metastasis to the fallopian tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Silvers, T.; Lawrence, S.; Kinders, R.; Parchment, R.; Teicher, B.A.; et al. 3D models of the NCI60 cell lines for screening oncology compounds. SLAS Discov. Adv. Life Sci. R D 2017, 22, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, S.M.; Karthikeyan, S.; Eddie, S.L.; Lantvit, D.D.; Ó hAinmhire, E.; Modi, D.A.; Wei, J.-J.; Burdette, J.E. Mutant p53 expression in fallopian tube epithelium drives cell migration. Int. J. Cancer 2015, 137, 1528–1538. [Google Scholar] [CrossRef]
- Pease, J.C.; Brewer, M.; Tirnauer, J.S. Spontaneous spheroid budding from monolayers: A potential contribution to ovarian cancer dissemination. Biol. Open 2012, 1, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.T.; Choi, K.-C.; Kang, S.K.; Leung, P.C.K. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102. [Google Scholar] [CrossRef]
- King, S.M.; Hilliard, T.S.; Wu, L.Y.; Jaffe, R.C.; Fazleabas, A.T.; Burdette, J.E. The impact of ovulation on fallopian tube epithelial cells: Evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr. Relat. Cancer 2011, 18, 627–642. [Google Scholar] [CrossRef]
- Huang, H.-S.; Chu, S.-C.; Hsu, C.-F.; Chen, P.-C.; Ding, D.-C.; Chang, M.-Y.; Chu, T.-Y. Mutagenic, surviving and tumorigenic effects of follicular fluid in the context of p53 loss: Initiation of fimbria. Carcinogenesis 2015, 36, 1419–1428. [Google Scholar] [CrossRef]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef]
- Saito, M.; Okamoto, A.; Kohno, T.; Takakura, S.; Shinozaki, H.; Isonishi, S.; Yasuhara, T.; Yoshimura, T.; Ohtake, Y.; Ochiai, K.; et al. Allelic imbalance and mutations of the PTEN gene in ovarian cancer. Int. J. Cancer 2000, 85, 160–165. [Google Scholar] [CrossRef]
- Lou, Y.; Yang, X.; Wang, F.; Cui, Z.; Huang, Y. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int. J. Mol. Med. 2010, 26, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.-J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.H.; Yassin, Y.; Miron, A.; Mehra, K.K.; Mehrad, M.; Monte, N.M.; Mutter, G.L.; Nucci, M.R.; Ning, G.; Mckeon, F.D.; et al. High-grade fimbrial-ovarian carcinomas are unified by altered p53, PTEN and PAX2 expression. Mod. Pathol. 2010, 23, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; de Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Pejovic, T.; Lawson, M.; Jurevic, L.; Hobbs, T.; Stouffer, R.L. Ovulation in the absence of the ovarian surface epithelium in the primate. Biol. Reprod. 2010, 82, 599–605. [Google Scholar] [CrossRef][Green Version]
- Wright, J.W.; Jurevic, L.; Stouffer, R.L. Dynamics of the primate ovarian surface epithelium during the ovulatory menstrual cycle. Hum. Reprod. 2011, 26, 1408–1421. [Google Scholar] [CrossRef]
- Merritt, M.A.; Pari, M.D.; Vitonis, A.F.; Titus, L.J.; Cramer, D.W.; Terry, K.L. Reproductive characteristics in relation to ovarian cancer risk by histologic pathways. Hum. Reprod. 2013, 28, 1406–1417. [Google Scholar] [CrossRef]
- Karst, A.M.; Levanon, K.; Drapkin, R. Modeling high-grade serous ovarian carcinogenesis from the fallopian tube. Proc. Natl. Acad. Sci. USA 2011, 108, 7547–7552. [Google Scholar] [CrossRef] [PubMed]
- Moyle-Heyrman, G.; Schipma, M.J.; Dean, M.; Davis, D.A.; Burdette, J.E. Genome-wide transcriptional regulation of estrogen receptor targets in fallopian tube cells and the role of selective estrogen receptor modulators. J. Ovarian Res. 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, L.H.; Ó hAinmhire, E.; Young, A.N.; Burdette, J.E.; Rodgers, L.H.; Ó hAinmhire, E.; Young, A.N.; Burdette, J.E. Loss of PAX8 in high-grade serous ovarian cancer reduces cell survival despite unique modes of action in the fallopian tube and ovarian surface epithelium. Oncotarget 2016, 7, 32785–32795. [Google Scholar] [CrossRef] [PubMed]
- Davidowitz, R.A.; Iwanicki, M.P.; Brugge, J.S. In vitro mesothelial clearance assay that models the early steps of ovarian cancer metastasis. J. Vis. Exp. 2012, 60, e3888. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Quartuccio, S.; Hilliard, T.S.; Inoue, K.; Burdette, J.E. Alginate hydrogels for three-dimensional organ culture of ovaries and oviducts. J. Vis. Exp. 2011, 52, 2804. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.; Souabni, A.; Busslinger, M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 2004, 38, 105–109. [Google Scholar] [CrossRef]
- Lewellen, K.A.; Metzinger, M.N.; Liu, Y.; Stack, M.S. Quantitation of intra-peritoneal ovarian cancer metastasis. J. Vis. Exp. 2016, 113, e53316. [Google Scholar] [CrossRef] [PubMed]
- Young, A.N.; Herrera, D.; Huntsman, A.C.; Korkmaz, M.A.; Lantvit, D.D.; Mazumder, S.; Kolli, S.; Coss, C.C.; King, S.; Wang, H.; et al. Phyllanthusmin derivatives induce apoptosis and reduce tumor burden in high-grade serous ovarian cancer by late-stage autophagy inhibition. Mol. Cancer Ther. 2018, 17, 2123–2135. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dean, M.; Jin, V.; Bergsten, T.M.; Austin, J.R.; Lantvit, D.D.; Russo, A.; Burdette, J.E. Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers 2019, 11, 884. https://doi.org/10.3390/cancers11060884
Dean M, Jin V, Bergsten TM, Austin JR, Lantvit DD, Russo A, Burdette JE. Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers. 2019; 11(6):884. https://doi.org/10.3390/cancers11060884
Chicago/Turabian StyleDean, Matthew, Vivian Jin, Tova M. Bergsten, Julia R. Austin, Daniel D. Lantvit, Angela Russo, and Joanna E. Burdette. 2019. "Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary" Cancers 11, no. 6: 884. https://doi.org/10.3390/cancers11060884
APA StyleDean, M., Jin, V., Bergsten, T. M., Austin, J. R., Lantvit, D. D., Russo, A., & Burdette, J. E. (2019). Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers, 11(6), 884. https://doi.org/10.3390/cancers11060884