Integrins, CAFs and Mechanical Forces in the Progression of Cancer


Abstract
1. Introduction
2. Integrins
2.1. Integrin Activation
2.2. Ligand Specificity and Diversity
2.3. Integrins in Mechanotransduction
3. Cancer-Associated Fibroblasts (CAFs)
CAF Activation
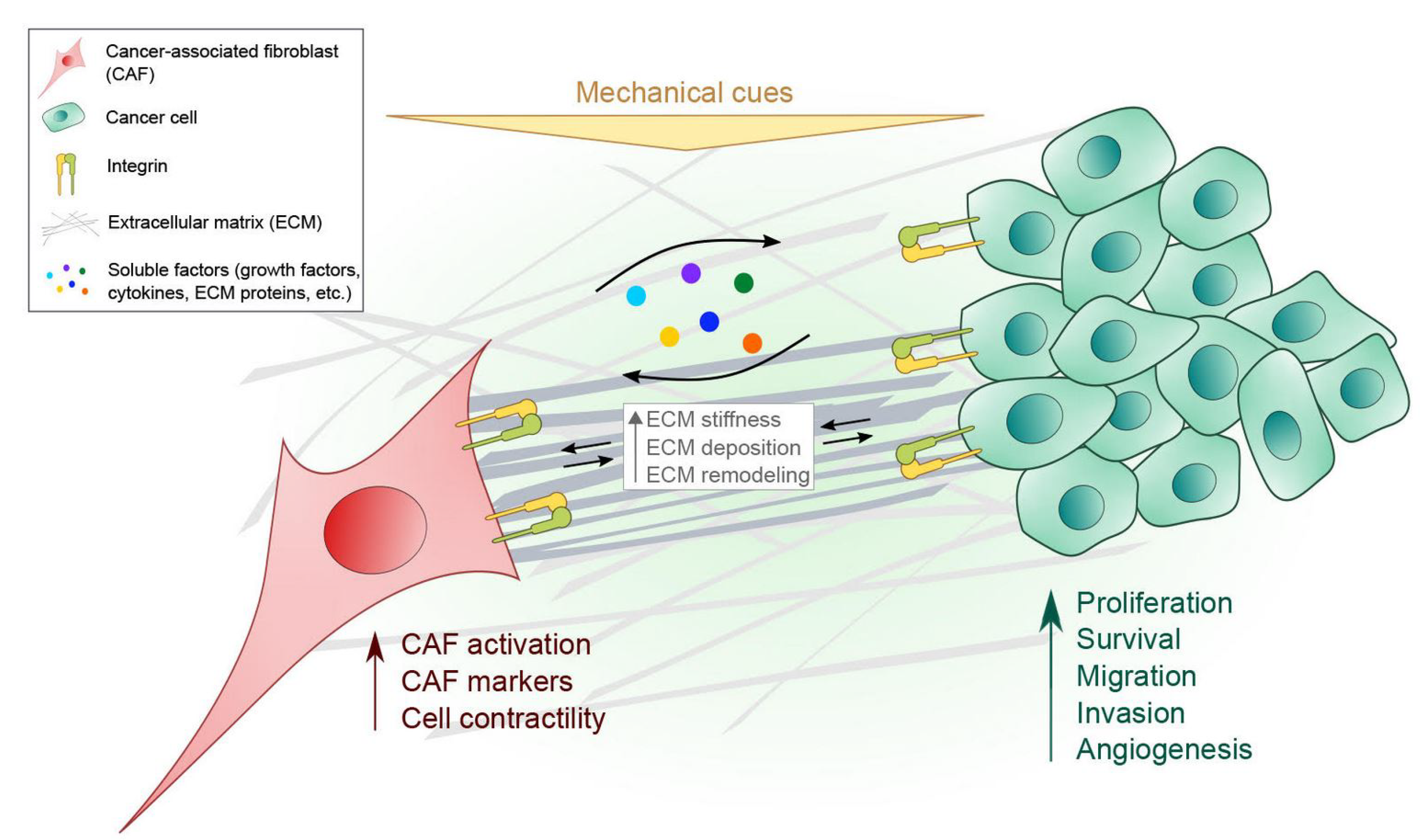
4. Communication Between CAFs and Cancer Cells Mediated by Integrins
4.1. ECM Remodeling by CAFs
4.2. Paracrine Signaling by CAFs to Cancer Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef]
- Bukoreshtliev, N.V.; Haase, K.; Pelling, A.E. Mechanical cues in cellular signalling and communication. Cell Tissue Res. 2013, 352, 77–94. [Google Scholar] [CrossRef]
- Chen, C.S.; Tan, J.; Tien, J. Mechanotransduction at cell-matrix and cell-cell contacts. Annu. Rev. Biomed. Eng. 2004, 6, 275–302. [Google Scholar] [CrossRef] [PubMed]
- Jaalouk, D.E.; Lammerding, J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 2009, 10, 63–73. [Google Scholar] [CrossRef]
- Orr, A.W.; Helmke, B.P.; Blackman, B.R.; Schwartz, M.A. Mechanisms of mechanotransduction. Dev. Cell 2006, 10, 11–20. [Google Scholar] [CrossRef]
- Gasparski, A.N.; Beningo, K.A. Mechanoreception at the cell membrane: More than the integrins. Arch. Biochem. Biophys. 2015, 586, 20–26. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Integrin diversity brings specificity in mechanotransduction. Biol. Cell 2018, 110, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Sporn, M.B. The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer 2007, 7, 139–147. [Google Scholar] [CrossRef]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef]
- Rasanen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes. Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Bouaouina, M.; Calderwood, D.A. Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 2009, 284, 11485–11497. [Google Scholar] [CrossRef]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin beta tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Puklin-Faucher, E.; Gao, M.; Schulten, K.; Vogel, V. How the headpiece hinge angle is opened: New insights into the dynamics of integrin activation. J. Cell Biol. 2006, 175, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Cheah, M.; Andrews, M.R. Integrin Activation: Implications for Axon Regeneration. Cells 2018, 7. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Morton, L.F.; Peachey, A.R.; Tuckwell, D.S.; Farndale, R.W.; Barnes, M.J. The collagen-binding A-domains of integrins α1β1 and α2β1 recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J. Biol. Chem. 2000, 275, 35–40. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Su, Y.; Chen, X.; Yu, L.; Mont, S.; Chetyrkin, S.; Voziyan, P.; Hudson, B.G.; Billings, P.C.; Jo, H.; et al. Inhibition of integrin α2β1 ameliorates glomerular injury. J. Am. Soc. Nephrol. 2012, 23, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Moeckel, G.; Morrow, J.D.; Cosgrove, D.; Harris, R.C.; Fogo, A.B.; Zent, R.; Pozzi, A. Lack of integrin α1β1 leads to severe glomerulosclerosis after glomerular injury. Am. J. Pathol. 2004, 165, 617–630. [Google Scholar] [CrossRef]
- Sutherland, M.; Gordon, A.; Shnyder, S.D.; Patterson, L.H.; Sheldrake, H.M. RGD-Binding Integrins in Prostate Cancer: Expression Patterns and Therapeutic Prospects against Bone Metastasis. Cancers 2012, 4, 1106–1145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.C. Integrin αvβ3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr. Drug Targets 2003, 4, 123–131. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Debordeaux, F.; Chansel-Debordeaux, L.; Pinaquy, J.B.; Fernandez, P.; Schulz, J. What about αvβ3 integrins in molecular imaging in oncology? Nucl. Med. Biol. 2018, 62–63, 31–46. [Google Scholar] [CrossRef]
- Hosotani, R.; Kawaguchi, M.; Masui, T.; Koshiba, T.; Ida, J.; Fujimoto, K.; Wada, M.; Doi, R.; Imamura, M. Expression of integrin αvβ3 in pancreatic carcinoma: Relation to MMP-2 activation and lymph node metastasis. Pancreas 2002, 25, e30–e35. [Google Scholar] [CrossRef]
- McCabe, N.P.; De, S.; Vasanji, A.; Brainard, J.; Byzova, T.V. Prostate cancer specific integrin αvβ3 modulates bone metastatic growth and tissue remodeling. Oncogene 2007, 26, 6238–6243. [Google Scholar] [CrossRef]
- Nip, J.; Shibata, H.; Loskutoff, D.J.; Cheresh, D.A.; Brodt, P. Human melanoma cells derived from lymphatic metastases use integrin αvβ3 to adhere to lymph node vitronectin. J. Clin. Investig. 1992, 90, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ishii, S.; Ikeda, T.; Masamura, S.; Doi, M.; Kitajima, M. The relationship between bone metastasis from human breast cancer and integrin αvβ3 expression. Anticancer Res. 2005, 25, 79–83. [Google Scholar] [PubMed]
- Hurt, E.M.; Chan, K.; Serrat, M.A.; Thomas, S.B.; Veenstra, T.D.; Farrar, W.L. Identification of vitronectin as an extrinsic inducer of cancer stem cell differentiation and tumor formation. Stem Cells 2010, 28, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Francolini, M.; Marthyn, P.; Zhang, J.; Carroll, R.S.; Nikas, D.C.; Strasser, J.F.; Villani, R.; Cheresh, D.A.; Black, P.M. αvβ3 and αvβ5 integrin expression in glioma periphery. Neurosurgery 2001, 49, 380–389. [Google Scholar] [PubMed]
- Felding-Habermann, B.; O'Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-beta3-dependent fibronectin assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Ouyang, G.; Bai, X.; Huang, Z.; Ma, C.; Liu, M.; Shao, R.; Anderson, R.M.; Rich, J.N.; Wang, X.F. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 2004, 5, 329–339. [Google Scholar] [CrossRef]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for αVβ3 and αVβ5 integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar]
- Shao, R.; Bao, S.; Bai, X.; Blanchette, C.; Anderson, R.M.; Dang, T.; Gishizky, M.L.; Marks, J.R.; Wang, X.F. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol. Cell. Biol. 2004, 24, 3992–4003. [Google Scholar] [CrossRef]
- Siriwardena, B.S.; Kudo, Y.; Ogawa, I.; Kitagawa, M.; Kitajima, S.; Hatano, H.; Tilakaratne, W.M.; Miyauchi, M.; Takata, T. Periostin is frequently overexpressed and enhances invasion and angiogenesis in oral cancer. Br. J. Cancer 2006, 95, 1396–1403. [Google Scholar] [CrossRef]
- Costanza, B.; Rademaker, G.; Tiamiou, A.; De Tullio, P.; Leenders, J.; Blomme, A.; Bellier, J.; Bianchi, E.; Turtoi, A.; Delvenne, P.; et al. Transforming growth factor beta-induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Franco-Barraza, J.; Francescone, R.; Luong, T.; Shah, N.; Madhani, R.; Cukierman, G.; Dulaimi, E.; Devarajan, K.; Egleston, B.L.; Nicolas, E.; et al. Matrix-regulated integrin αVβ5 maintains α5β1-dependent desmoplastic traits prognostic of neoplastic recurrence. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zou, X.; Xia, W.; Gao, H.; Li, Z.; Liu, N.; Xu, Z.; Gao, C.; He, Z.; Niu, W.; et al. Integrin αVβ6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Radwanska, A.; Beghelli-de la Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The Interaction between Laminin-332 and α3β1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, J.; Choi, C.; Kim, J.H. Blockade of integrin α3 attenuates human pancreatic cancer via inhibition of EGFR signalling. Sci. Rep. 2019, 9, 2793. [Google Scholar] [CrossRef]
- Lusche, D.F.; Klemme, M.R.; Soll, B.A.; Reis, R.J.; Forrest, C.C.; Nop, T.S.; Wessels, D.J.; Berger, B.; Glover, R.; Soll, D.R. Integrin α-3 ß-1’s central role in breast cancer, melanoma and glioblastoma cell aggregation revealed by antibodies with blocking activity. MAbs 2019. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Miyazaki, K.; Oyanagi, J.; Hoshino, D.; Togo, S.; Kumagai, H.; Miyagi, Y. Cancer cell migration on elongate protrusions of fibroblasts in collagen matrix. Sci. Rep. 2019, 9, 292. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef]
- Wang, K.K.; Liu, N.; Radulovich, N.; Wigle, D.A.; Johnston, M.R.; Shepherd, F.A.; Minden, M.D.; Tsao, M.S. Novel candidate tumor marker genes for lung adenocarcinoma. Oncogene 2002, 21, 7598–7604. [Google Scholar] [CrossRef]
- Zhu, C.Q.; Popova, S.N.; Brown, E.R.; Barsyte-Lovejoy, D.; Navab, R.; Shih, W.; Li, M.; Lu, M.; Jurisica, I.; Penn, L.Z.; et al. Integrin α11 regulates IGF2 expression in fibroblasts to enhance tumorigenicity of human non-small-cell lung cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 11754–11759. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Mardhian, D.F.; van Baarlen, J.; Ostman, A.; Prakash, J. Integrin α11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; McMillan, R.H.; Chang, Y.T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions with Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Q.; Yu, Z.; Wu, X.; Chen, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; Liu, B.; et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin beta1-FAK signaling pathway. Int. J. Cancer 2017, 141, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Gasparski, A.N.; Ozarkar, S.; Beningo, K.A. Transient mechanical strain promotes the maturation of invadopodia and enhances cancer cell invasion in vitro. J. Cell Sci. 2017, 130, 1965–1978. [Google Scholar] [CrossRef]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin beta3-p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef]
- Samani, A.; Zubovits, J.; Plewes, D. Elastic moduli of normal and pathological human breast tissues: An inversion-technique-based investigation of 169 samples. Phys. Med. Biol. 2007, 52, 1565–1576. [Google Scholar] [CrossRef]
- Tung, J.C.; Barnes, J.M.; Desai, S.R.; Sistrunk, C.; Conklin, M.W.; Schedin, P.; Eliceiri, K.W.; Keely, P.J.; Seewaldt, V.L.; Weaver, V.M. Tumor mechanics and metabolic dysfunction. Free Radic. Biol. Med. 2015, 79, 269–280. [Google Scholar] [CrossRef]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef]
- Mierke, C.T.; Sauer, F.; Grosser, S.; Puder, S.; Fischer, T.; Kas, J.A. The two faces of enhanced stroma: Stroma acts as a tumor promoter and a steric obstacle. NMR Biomed. 2018, 31, e3831. [Google Scholar] [CrossRef]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef]
- Ladoux, B.; Mege, R.M.; Trepat, X. Front-Rear Polarization by Mechanical Cues: From Single Cells to Tissues. Trends Cell Biol. 2016, 26, 420–433. [Google Scholar] [CrossRef]
- Muhamed, I.; Chowdhury, F.; Maruthamuthu, V. Biophysical Tools to Study Cellular Mechanotransduction. Bioengineering 2017, 4. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Bazellieres, E.; Allen, M.D.; Andreu, I.; Oria, R.; Sunyer, R.; Gomm, J.J.; Marshall, J.F.; Jones, J.L.; Trepat, X.; et al. Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 2014, 13, 631–637. [Google Scholar] [CrossRef]
- Undyala, V.V.; Dembo, M.; Cembrola, K.; Perrin, B.J.; Huttenlocher, A.; Elce, J.S.; Greer, P.A.; Wang, Y.L.; Beningo, K.A. The calpain small subunit regulates cell-substrate mechanical interactions during fibroblast migration. J. Cell Sci. 2008, 121, 3581–3588. [Google Scholar] [CrossRef]
- Franco, S.J.; Huttenlocher, A. Regulating cell migration: Calpains make the cut. J. Cell Sci. 2005, 118, 3829–3838. [Google Scholar] [CrossRef]
- Sackmann, E. How actin/myosin crosstalks guide the adhesion, locomotion and polarization of cells. Biochim. Biophys. Acta 2015, 1853, 3132–3142. [Google Scholar] [CrossRef]
- Wells, A.; Huttenlocher, A.; Lauffenburger, D.A. Calpain proteases in cell adhesion and motility. Int. Rev. Cytol. 2005, 245, 1–16. [Google Scholar] [CrossRef]
- Indra, I.; Undyala, V.; Kandow, C.; Thirumurthi, U.; Dembo, M.; Beningo, K.A. An in vitro correlation of mechanical forces and metastatic capacity. Phys. Biol. 2011, 8, 015015. [Google Scholar] [CrossRef]
- Jiang, G.; Huang, A.H.; Cai, Y.; Tanase, M.; Sheetz, M.P. Rigidity sensing at the leading edge through αvβ3 integrins and RPTPα. Biophys. J. 2006, 90, 1804–1809. [Google Scholar] [CrossRef]
- Chen, W.; Lou, J.; Evans, E.A.; Zhu, C. Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J. Cell Biol. 2012, 199, 497–512. [Google Scholar] [CrossRef]
- Chen, Y.; Lee, H.; Tong, H.; Schwartz, M.; Zhu, C. Force regulated conformational change of integrin αVβ3. Matrix Biol. 2017, 60–61, 70–85. [Google Scholar] [CrossRef]
- Zhang, X.; Craig, S.E.; Kirby, H.; Humphries, M.J.; Moy, V.T. Molecular basis for the dynamic strength of the integrin α4β1/VCAM-1 interaction. Biophys. J. 2004, 87, 3470–3478. [Google Scholar] [CrossRef]
- Burgos-Bravo, F.; Figueroa, N.L.; Casanova-Morales, N.; Quest, A.F.G.; Wilson, C.A.M.; Leyton, L. Single-molecule measurements of the effect of force on Thy-1/αvβ3- integrin interaction using nonpurified proteins. Mol. Biol. Cell 2018, 29, 326–338. [Google Scholar] [CrossRef]
- Menon, S.; Beningo, K.A. Cancer cell invasion is enhanced by applied mechanical stimulation. PLoS ONE 2011, 6, e17277. [Google Scholar] [CrossRef]
- Chen, K.D.; Li, Y.S.; Kim, M.; Li, S.; Yuan, S.; Chien, S.; Shyy, J.Y. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and Shc. J. Biol. Chem. 1999, 274, 18393–18400. [Google Scholar] [CrossRef]
- Tzima, E.; del Pozo, M.A.; Shattil, S.J.; Chien, S.; Schwartz, M.A. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001, 20, 4639–4647. [Google Scholar] [CrossRef]
- Choi, Y.I.; Duke-Cohan, J.S.; Chen, W.; Liu, B.; Rossy, J.; Tabarin, T.; Ju, L.; Gui, J.; Gaus, K.; Zhu, C.; et al. Dynamic control of beta1 integrin adhesion by the plexinD1-sema3E axis. Proc. Natl. Acad. Sci. USA 2014, 111, 379–384. [Google Scholar] [CrossRef]
- Hinz, B.; Mastrangelo, D.; Iselin, C.E.; Chaponnier, C.; Gabbiani, G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 2001, 159, 1009–1020. [Google Scholar] [CrossRef]
- Hinz, B.; Gabbiani, G. Mechanisms of force generation and transmission by myofibroblasts. Curr. Opin. Biotechnol. 2003, 14, 538–546. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef]
- Amatangelo, M.D.; Bassi, D.E.; Klein-Szanto, A.J.; Cukierman, E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am. J. Pathol. 2005, 167, 475–488. [Google Scholar] [CrossRef]
- Hogdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Kalli, M.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Solid Stress Facilitates Fibroblasts Activation to Promote Pancreatic Cancer Cell Migration. Ann. Biomed. Eng. 2018, 46, 657–669. [Google Scholar] [CrossRef]
- Cheng, G.; Tse, J.; Jain, R.K.; Munn, L.L. Micro-environmental mechanical stress controls tumor spheroid size and morphology by suppressing proliferation and inducing apoptosis in cancer cells. PLoS ONE 2009, 4, e4632. [Google Scholar] [CrossRef]
- Delarue, M.; Montel, F.; Vignjevic, D.; Prost, J.; Joanny, J.F.; Cappello, G. Compressive stress inhibits proliferation in tumor spheroids through a volume limitation. Biophys. J. 2014, 107, 1821–1828. [Google Scholar] [CrossRef]
- Helmlinger, G.; Netti, P.A.; Lichtenbeld, H.C.; Melder, R.J.; Jain, R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997, 15, 778–783. [Google Scholar] [CrossRef]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar] [CrossRef]
- Givant-Horwitz, V.; Davidson, B.; Reich, R. Laminin-induced signaling in tumor cells. Cancer Lett. 2005, 223, 1–10. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar]
- Sun, X.; Mao, Y.; Wang, J.; Zu, L.; Hao, M.; Cheng, G.; Qu, Q.; Cui, D.; Keller, E.T.; Chen, X.; et al. IL-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene 2014. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef]
- Huang, W.; Chiquet-Ehrismann, R.; Moyano, J.V.; Garcia-Pardo, A.; Orend, G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001, 61, 8586–8594. [Google Scholar]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef]
- Yan, W.; Shao, R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J. Biol. Chem. 2006, 281, 19700–19708. [Google Scholar] [CrossRef]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef]
- Takayama, G.; Arima, K.; Kanaji, T.; Toda, S.; Tanaka, H.; Shoji, S.; McKenzie, A.N.; Nagai, H.; Hotokebuchi, T.; Izuhara, K. Periostin: A novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 2006, 118, 98–104. [Google Scholar] [CrossRef]
- Tian, B.; Chen, X.; Zhang, H.; Li, X.; Wang, J.; Han, W.; Zhang, L.Y.; Fu, L.; Li, Y.; Nie, C.; et al. Urokinase plasminogen activator secreted by cancer-associated fibroblasts induces tumor progression via PI3K/AKT and ERK signaling in esophageal squamous cell carcinoma. Oncotarget 2017, 8, 42300–42313. [Google Scholar] [CrossRef][Green Version]
- Ciavarella, S.; Laurenzana, A.; De Summa, S.; Pilato, B.; Chilla, A.; Lacalamita, R.; Minoia, C.; Margheri, F.; Iacobazzi, A.; Rana, A.; et al. u-PAR expression in cancer associated fibroblast: New acquisitions in multiple myeloma progression. BMC Cancer 2017, 17, 215. [Google Scholar] [CrossRef]
- Kawase, A.; Ishii, G.; Nagai, K.; Ito, T.; Nagano, T.; Murata, Y.; Hishida, T.; Nishimura, M.; Yoshida, J.; Suzuki, K.; et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int. J. Cancer 2008, 123, 1053–1059. [Google Scholar] [CrossRef]
- Pula, B.; Jethon, A.; Piotrowska, A.; Gomulkiewicz, A.; Owczarek, T.; Calik, J.; Wojnar, A.; Witkiewicz, W.; Rys, J.; Ugorski, M.; et al. Podoplanin expression by cancer-associated fibroblasts predicts poor outcome in invasive ductal breast carcinoma. Histopathology 2011, 59, 1249–1260. [Google Scholar] [CrossRef]
- Pula, B.; Witkiewicz, W.; Dziegiel, P.; Podhorska-Okolow, M. Significance of podoplanin expression in cancer-associated fibroblasts: A comprehensive review. Int. J. Oncol. 2013, 42, 1849–1857. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Jesch, B.; Riegler, M.F.; Maroske, F.; Schwameis, K.; Jomrich, G.; Birner, P. Podoplanin expressing cancer associated fibroblasts are associated with unfavourable prognosis in adenocarcinoma of the esophagus. Clin. Exp. Metastasis 2013, 30, 441–446. [Google Scholar] [CrossRef]
- Yamanashi, T.; Nakanishi, Y.; Fujii, G.; Akishima-Fukasawa, Y.; Moriya, Y.; Kanai, Y.; Watanabe, M.; Hirohashi, S. Podoplanin expression identified in stromal fibroblasts as a favorable prognostic marker in patients with colorectal carcinoma. Oncology 2009, 77, 53–62. [Google Scholar] [CrossRef]
- Yurugi, Y.; Wakahara, M.; Matsuoka, Y.; Sakabe, T.; Kubouchi, Y.; Haruki, T.; Nosaka, K.; Miwa, K.; Araki, K.; Taniguchi, Y.; et al. Podoplanin Expression in Cancer-associated Fibroblasts Predicts Poor Prognosis in Patients with Squamous Cell Carcinoma of the Lung. Anticancer Res. 2017, 37, 207–213. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Integrins | Source of Expression | Ligands in the Study | Cancer-Promoting Effects | References |
|---|---|---|---|---|
| αvβ3 | Prostate and breast CSC | Vitronectin | Tumor formation | [44] |
| Glioblastoma | Glioma-associated angiogenesis | [45] | ||
| Breast cancer | Shear stress | Metastatic potential | [46] | |
| CAF | Fibronectin | Colon cancer cell invasion | [47] | |
| CAF | Periostin | Cell survival, growth, proliferation, migration, invasion and angiogenesis | [48,49,50,51] | |
| αvβ5 | Glioblastoma | Glioma-associated angiogenesis | [45] | |
| Pancreatic cancer | TGFBI | Pancreatic cancer cell invasion | [52] | |
| CAF | Differentiation of normal fibroblasts into CAFs | [53] | ||
| CAF | Periostin | Cell survival, growth, proliferation, migration, invasion and angiogenesis | [48,49,50,51] | |
| αvβ6 | CRC | CAF activation and CRC invasion | [54] | |
| HNSCC | Fibronectin | Directional migration of collective HNSCC cells | [55] | |
| α3β1 | CAF | Collective invasion of SCC cells | [56] | |
| CAF | Laminin-322 | CAF differentiation | [57] | |
| Pancreatic cancer | Poor clinical outcome in pancreatic cancer patients | [58] | ||
| Breast cancer, melanoma and glioblastoma | Cell aggregation and aggregate coalescence | [59] | ||
| α5β1 | CAF | Fibronectin | Differentiation of normal fibroblasts into CAFs | [53] |
| CAF | Collective invasion of SCC cells | [56] | ||
| CAF | Fibronectin | Integrin αv-mediated directional pancreatic cancer cell migration | [60] | |
| Pancreatic and lung cancer | Fibronectin | Direct interaction of cancer cells with fibronectin-presenting fibroblasts | [61] | |
| α9β1 | HNSCC | Fibronectin | Directional migration of collective HNSCC cells | [55] |
| α11β1 | CAF | Collagen | Non-small cell lung carcinoma progression | [62,63,64,65] |
| PSC | Collagen | Pancreatic cancer cell migration and invasion | [66] | |
| β1 | Pancreatic cancer | Collagen | Tumor initiation, self-renewal potential, migration and invasion | [67,68] |
| Pancreatic and breast cancer | Fibronectin | Pancreatic and breast cancer cell invasion | [69] | |
| Gastric cancer | Tumorigenesis and metastasis of gastric cancer | [70] | ||
| β3 | Fibrosarcoma | Transient tugging and pulling forces | Integrin β3 downregulation-mediated fibrosarcoma cell invasion | [71] |
| Breast cancer | IL-32 | Breast cancer cell invasion and metastasis | [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. https://doi.org/10.3390/cancers11050721
Jang I, Beningo KA. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers. 2019; 11(5):721. https://doi.org/10.3390/cancers11050721
Chicago/Turabian StyleJang, Imjoo, and Karen A. Beningo. 2019. "Integrins, CAFs and Mechanical Forces in the Progression of Cancer" Cancers 11, no. 5: 721. https://doi.org/10.3390/cancers11050721
APA StyleJang, I., & Beningo, K. A. (2019). Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers, 11(5), 721. https://doi.org/10.3390/cancers11050721