Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Generation of ER+ and ER− Palbociclib-Resistant Breast Cancer Cell Models
2.2. Acquired Resistance to Palbociclib Promotes Sensitivity to OAdmCherry Only in ER+ MCF7 Breast Cancer Cells
2.3. Palbociclib Enhances OAdmCherry Replication and Oncolytic Properties Only in ER+ MCF7 Breast Cancer Cells
2.4. Coxsackie-Adenovirus Receptor (CAR) Expression Correlates with Replication and Oncolytic Potential of OAdmCherry in Palbociclib-Resistant Cells
2.5. Evaluation of Type I IFN Response in ER+ and ER− Palbociclib-Resistant Breast Cancer Cells
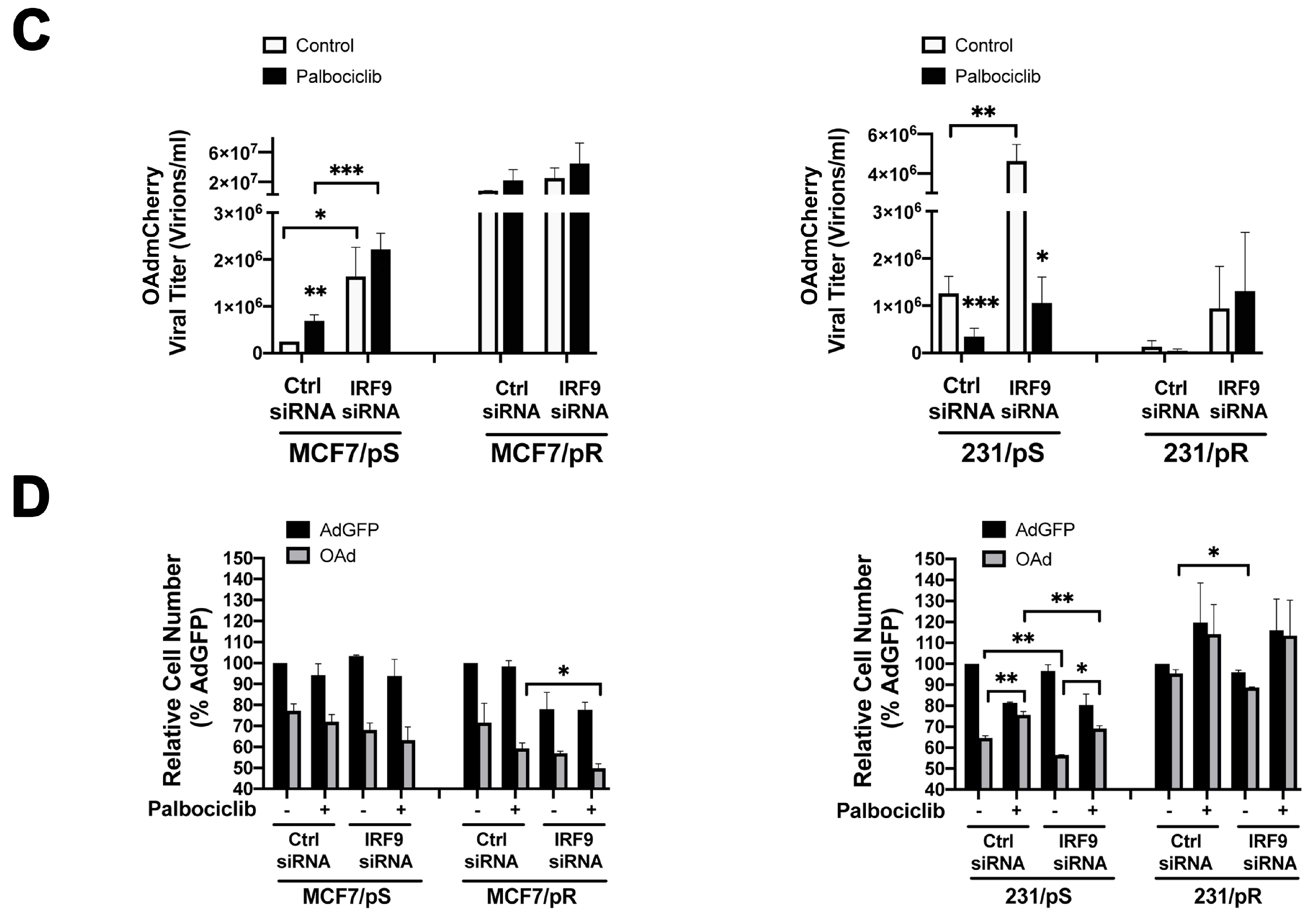
2.6. Downregulation of IRF9 Increases OAdmCherry Replication and Oncolysis in Palbociclib-Sensitive Breast Cancer Cells
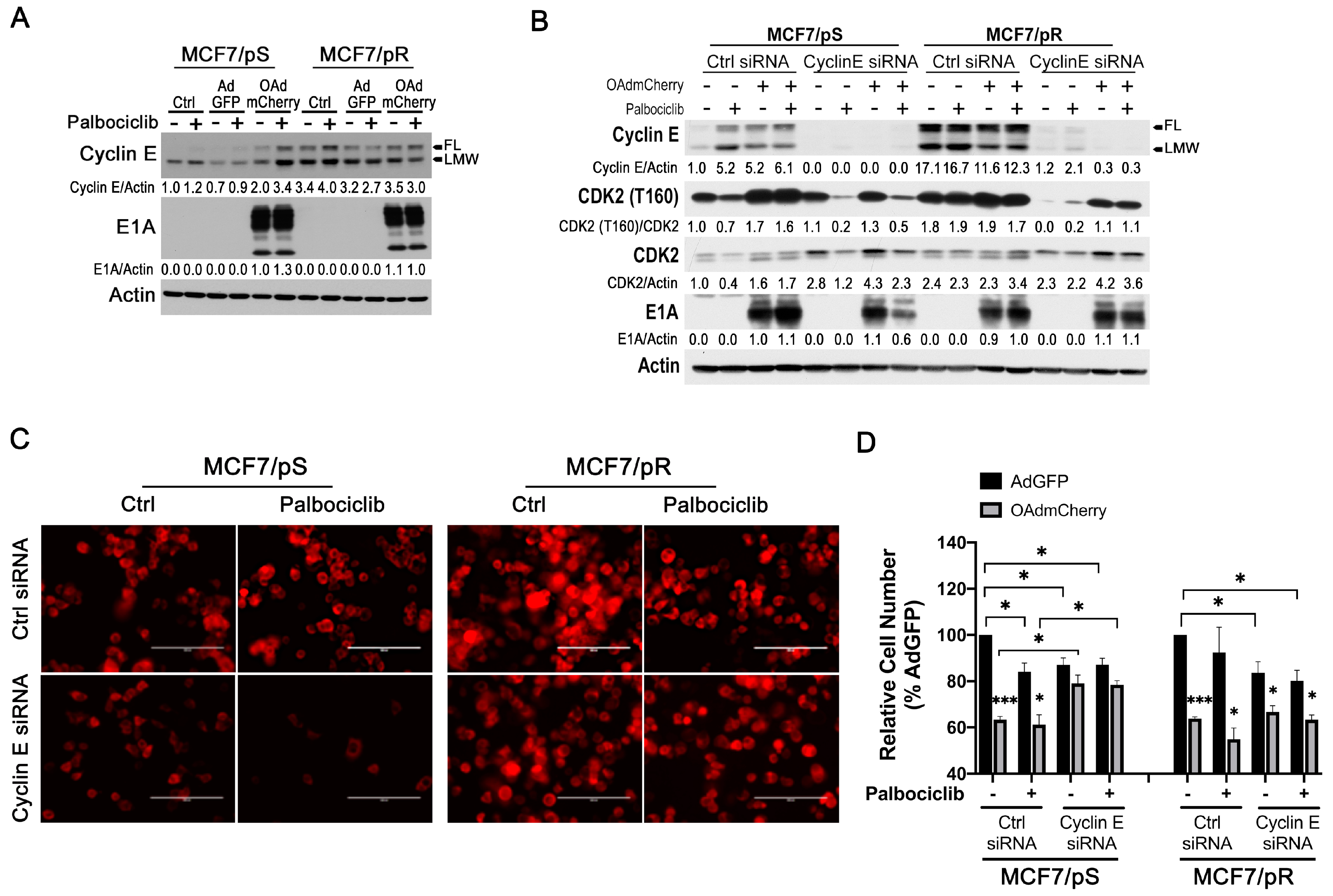
2.7. Cyclin E Knockdown Blocks Virus Replication Only in MCF7/pS Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture, Generation of Palbociclib-Resistant Cells and Palbociclib Treatment
4.3. Cell Proliferation Assays
4.4. Cell Cycle Analysis
4.5. RNA Extraction and Next-Generation Sequencing
4.6. Adenoviral Vectors
4.7. Combined Therapies
4.8. Adenovirus Titer Assay
4.9. Western Blot Analysis
4.10. siRNA Transfection
4.11. Fluorescence Microscopy
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Scott, S.C.; Lee, S.S.; Abraham, J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin. Oncol. 2017, 44, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine therapy for hormone receptor-positive metastatic breast cancer: american society of clinical oncology guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Aromatase inhibitors versus tamoxifen in early breast cancer: Patient-level meta-analysis of the randomised trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Dickler, M.N. Estrogen receptor-positive breast cancer: exploiting signaling pathways implicated in endocrine resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef]
- Hui, R.; Finney, G.L.; Carroll, J.S.; Lee, C.S.; Musgrove, E.A.; Sutherland, R.L. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002, 62, 6916–6923. [Google Scholar]
- Stendahl, M.; Kronblad, A.; Ryden, L.; Emdin, S.; Bengtsson, N.O.; Landberg, G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br. J. Cancer 2004, 90, 1942–1948. [Google Scholar] [CrossRef]
- Jirstrom, K.; Stendahl, M.; Ryden, L.; Kronblad, A.; Bendahl, P.O.; Stal, O.; Landberg, G. Adverse effect of adjuvant tamoxifen in premenopausal breast cancer with cyclin D1 gene amplification. Cancer Res. 2005, 65, 8009–8016. [Google Scholar] [CrossRef]
- Rudas, M.; Lehnert, M.; Huynh, A.; Jakesz, R.; Singer, C.; Lax, S.; Schippinger, W.; Dietze, O.; Greil, R.; Stiglbauer, W.; et al. Cyclin D1 expression in breast cancer patients receiving adjuvant tamoxifen-based therapy. Clin. Cancer Res. 2008, 14, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; LoRusso, P.M.; Dickson, M.A.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; Courtney, R.; O’Dwyer, P.J. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br. J. Cancer 2011, 104, 1862–1868. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular pathways: Targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef]
- Dhillon, S. Palbociclib: First global approval. Drugs 2015, 75, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sanchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T.; et al. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat. Commun. 2017, 8, 13923. [Google Scholar] [CrossRef]
- Liu, T.C.; Thorne, S.H.; Kirn, D.H. Oncolytic adenoviruses for cancer gene therapy. Methods Mol. Biol. 2008, 433, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Sampson-Johannes, A.; Williams, A.; McCormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Natl. Med. 1997, 3, 639–645. [Google Scholar] [CrossRef]
- Xia, Z.J.; Chang, J.H.; Zhang, L.; Jiang, W.Q.; Guan, Z.Z.; Liu, J.W.; Zhang, Y.; Hu, X.H.; Wu, G.H.; Wang, H.Q.; et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng 2004, 23, 1666–1670. [Google Scholar] [PubMed]
- Heise, C.; Lemmon, M.; Kirn, D. Efficacy with a replication-selective adenovirus plus cisplatin-based chemotherapy: Dependence on sequencing but not p53 functional status or route of administration. Clin. Cancer Res. 2000, 6, 4908–4914. [Google Scholar]
- Galanis, E.; Okuno, S.H.; Nascimento, A.G.; Lewis, B.D.; Lee, R.A.; Oliveira, A.M.; Sloan, J.A.; Atherton, P.; Edmonson, J.H.; Erlichman, C.; et al. Phase I-II trial of ONYX-015 in combination with MAP chemotherapy in patients with advanced sarcomas. Gene Ther. 2005, 12, 437–445. [Google Scholar] [CrossRef]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Monsurro, V.; Beghelli, S.; Wang, R.; Barbi, S.; Coin, S.; Di Pasquale, G.; Bersani, S.; Castellucci, M.; Sorio, C.; Eleuteri, S.; et al. Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: Potential relevance for adenoviral gene therapy. J. Transl. Med. 2010, 8, 10. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Shah, N.; Murphy, A.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V. Resistance of Pancreatic Cancer Cells to Oncolytic Vesicular Stomatitis Virus: Role of Type I Interferon Signaling; Elsevier: Amsterdam, The Netherlands, 2012; Volume 436. [Google Scholar]
- Liikanen, I.; Monsurro, V.; Ahtiainen, L.; Raki, M.; Hakkarainen, T.; Diaconu, I.; Escutenaire, S.; Hemminki, O.; Dias, J.D.; Cerullo, V.; et al. Induction of interferon pathways mediates in vivo resistance to oncolytic adenovirus. Mol. Ther. 2011, 19, 1858–1866. [Google Scholar] [CrossRef]
- Danziger, O.; Pupko, T.; Bacharach, E.; Ehrlich, M. Interleukin-6 and Interferon-alpha Signaling via JAK1-STAT Differentially Regulate Oncolytic versus Cytoprotective Antiviral States. Front. Immunol. 2018, 9, 94. [Google Scholar] [CrossRef]
- Zheng, X.; Rao, X.M.; Gomez-Gutierrez, J.G.; Hao, H.; McMasters, K.M.; Zhou, H.S. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J. Virol. 2008, 82, 3415–3427. [Google Scholar] [CrossRef]
- Cheng, P.H.; Rao, X.M.; McMasters, K.M.; Zhou, H.S. Molecular basis for viral selective replication in cancer cells: Activation of CDK2 by adenovirus-induced cyclin E. PLoS ONE 2013, 8, e57340. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; Andre, F.; Bayar, M.A.; et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Teh, J.L.F.; Aplin, A.E. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Garza-Morales, R.; Gonzalez-Ramos, R.; Chiba, A.; Montes de Oca-Luna, R.; McNally, L.R.; McMasters, K.M.; Gomez-Gutierrez, J.G. Temozolomide enhances triple-negative breast cancer virotherapy in vitro. Cancers (Basel) 2018, 10, 144. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT 2013, 2, e23931. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Leonard, G.T.; Sen, G.C. Effects of adenovirus E1A protein on interferon-signaling. Virology 1996, 224, 25–33. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet. Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Myrand, S.P.; et al. LY2835219, a novel cell cycle inhibitor selective for CDK4/6, in combination with fulvestrant for patients with hormone receptor positive (HR plus) metastatic breast cancer. J. Clin. Oncol. 2014, 32, 534. [Google Scholar] [CrossRef]
- Infante, J.R.; Shapiro, G.; Witteveen, P.; Gerecitano, J.F.; Ribrag, V.; Chugh, R.; Issa, I.; Chakraborty, A.; Matano, A.; Zhao, X.M.; et al. A phase I study of the single-agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas. J. Clin. Oncol. 2014, 32, 2528. [Google Scholar] [CrossRef]
- Shapiro, G.; Rosen, L.S.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Tolaney, S.M.; Beeram, M.; Rasco, D.W.; Kulanthaivel, P.; et al. A first-in-human phase I study of the CDK4/6 inhibitor, LY2835219, for patients with advanced cancer. J. Clin. Oncol. 2013, 31, 2500. [Google Scholar]
- Pandey, K.; An, H.J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.M.; Silk, A.W.; Zloza, A. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front. Oncol. 2017, 7, 202. [Google Scholar] [CrossRef]
- Lundstrom, K. New frontiers in oncolytic viruses: Optimizing and selecting for virus strains with improved efficacy. Biologics 2018, 12, 43–60. [Google Scholar] [CrossRef]
- Suryawanshi, Y.R.; Zhang, T.; Essani, K. Oncolytic viruses: Emerging options for the treatment of breast cancer. Med. Oncol. 2017, 34, 43. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L.; et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol. Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef]
- Oberg, D.; Yanover, E.; Adam, V.; Sweeney, K.; Costas, C.; Lemoine, N.R.; Hallden, G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin. Cancer Res. 2010, 16, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Harding, B.; Aspuria, P.J.; Agadjanian, H.; Cheon, D.J.; Mizuno, T.; Greenberg, D.; Allen, J.R.; Spurka, L.; Funari, V.; Spiteri, E.; et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015, 6, 696–714. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. NeoPalAna: Neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor-positive breast cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Goel, S. CDK4/6 inhibition in breast cancer: Mechanisms of response and treatment failure. Curr. Breast Cancer Rep. 2017, 9, 26–33. [Google Scholar] [CrossRef]
- Walter, K.R.; Goodman, M.L.; Singhal, H.; Hall, J.A.; Li, T.; Holloran, S.M.; Trinca, G.M.; Gibson, K.A.; Jin, V.X.; Greene, G.L.; et al. Interferon-stimulated genes are transcriptionally repressed by PR in breast cancer. Mol. Cancer Res. MCR 2017, 15, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.L.; Trinca, G.M.; Walter, K.R.; Papachristou, E.K.; D’Santos, C.S.; Li, T.; Liu, Q.; Lai, Z.; Chalise, P.; Madan, R.; et al. Progesterone receptor attenuates STAT1-mediated IFN signaling in breast cancer. J. Immunol. 2019, 202, 3076–3086. [Google Scholar] [CrossRef]
- Maia, C.; Rocha, S.; Socorro, S.; Schmitt, F.; Santos, C. Oligodenylate synthetase 1 (oas1) expression in human breast and prostate cancer cases, and its regulation by sex steroid hormones. Adv. Mod. Oncol. Res. 2016, 2, 97–110. [Google Scholar] [CrossRef][Green Version]
- Illumina Proprietary. BaseSpace User Guide; Illumina: San Diego, CA, USA, 2014; Volume 15044182, Review E; pp. 1–98. [Google Scholar]
- Egger, M.E.; McNally, L.R.; Nitz, J.; McMasters, K.M.; Gomez-Gutierrez, J.G. Adenovirus-mediated FKHRL1/TM sensitizes melanoma cells to apoptosis induced by temozolomide. Hum. Gene Ther. Clin. Dev. 2014, 25, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Borovjagin, A.V.; McNally, L.R.; Wang, M.; Curiel, D.T.; MacDougall, M.J.; Zinn, K.R. Noninvasive monitoring of mRFP1- and mCherry-labeled oncolytic adenoviruses in an orthotopic breast cancer model by spectral imaging. Mol. Imaging 2010, 9, 59–75. [Google Scholar] [CrossRef]
- Wechman, S.L.; Rao, X.M.; Cheng, P.H.; Gomez-Gutierrez, J.G.; McMasters, K.M.; Zhou, H.S. Development of an oncolytic adenovirus with enhanced spread ability through repeated uv irradiation and cancer selection. Viruses 2016, 8, 167. [Google Scholar] [CrossRef]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lypova, N.; Lanceta, L.; Gipson, A.; Vega, S.; Garza-Morales, R.; McMasters, K.M.; Chesney, J.; Gomez-Gutierrez, J.G.; Imbert-Fernandez, Y. Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy. Cancers 2019, 11, 684. https://doi.org/10.3390/cancers11050684
Lypova N, Lanceta L, Gipson A, Vega S, Garza-Morales R, McMasters KM, Chesney J, Gomez-Gutierrez JG, Imbert-Fernandez Y. Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy. Cancers. 2019; 11(5):684. https://doi.org/10.3390/cancers11050684
Chicago/Turabian StyleLypova, Nadiia, Lilibeth Lanceta, Alana Gipson, Stephanie Vega, Rodolfo Garza-Morales, Kelly M. McMasters, Jason Chesney, Jorge G. Gomez-Gutierrez, and Yoannis Imbert-Fernandez. 2019. "Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy" Cancers 11, no. 5: 684. https://doi.org/10.3390/cancers11050684
APA StyleLypova, N., Lanceta, L., Gipson, A., Vega, S., Garza-Morales, R., McMasters, K. M., Chesney, J., Gomez-Gutierrez, J. G., & Imbert-Fernandez, Y. (2019). Targeting Palbociclib-Resistant Estrogen Receptor-Positive Breast Cancer Cells via Oncolytic Virotherapy. Cancers, 11(5), 684. https://doi.org/10.3390/cancers11050684