Abstract
Pheochromocytomas and paragangliomas (PPGL) are rare neuroendocrine tumors that show the highest heritability of all human neoplasms and represent a paradoxical example of genetic heterogeneity. Amongst the elevated number of genes involved in the hereditary predisposition to the disease (at least nineteen) there are eleven tricarboxylic acid (TCA) cycle-related genes, some of which are also involved in the development of congenital recessive neurological disorders and other cancers such as cutaneous and uterine leiomyomas, gastrointestinal tumors and renal cancer. Somatic or germline mutation of genes encoding enzymes catalyzing pivotal steps of the TCA cycle not only disrupts cellular respiration, but also causes severe alterations in mitochondrial metabolite pools. These latter alterations lead to aberrant accumulation of “oncometabolites” that, in the end, may lead to deregulation of the metabolic adaptation of cells to hypoxia, inhibition of the DNA repair processes and overall pathological changes in gene expression. In this review, we will address the TCA cycle mutations leading to the development of PPGL, and we will discuss the relevance of these mutations for the transformation of neural crest-derived cells and potential therapeutic approaches based on the emerging knowledge of underlying molecular alterations.
1. Metabolism and Cancer
Almost a century ago, Nobel Prize winner Otto Warburg described how cancer cells can reprogram glucose metabolism by dramatically increasing the rate of glucose uptake, which is fermented to produce lactate even in the presence of oxygen and fully functioning mitochondria [1]. This observation suggested that defects in mitochondrial respiration could be the underlying cause of cancer. Although this aerobic glycolytic mechanism, known as the Warburg effect, has been studied extensively, its benefits for cell growth and survival are not well understood. In fact, nowadays it is more accepted that genetic events occurring in cancer cells are the cause of the alterations in metabolism observed by Warburg in the 1920s. At the beginning of the 21st century, the first mutations in the SDHD gene were reported, providing for the first time a link between germline alterations in a metabolic gene and the development of cancer, and demonstrating how disruption of mitochondrial respiration may lead to tumor development [2]. Moreover, the description of the first germline mutations in the SDHD gene in patients with hereditary pheochromocytoma (PCC) and paraganglioma (PGL) (together referred to as PPGL) marked a milestone in the study of this rare disease.
2. Germline or Somatic Disruption of the Tricarboxylic Acid (TCA) Cycle Leads to PPGL Development
For a long time it was thought that the tricarboxylic acid (TCA) cycle was so crucial to the metabolism of living cells that any significant defect, including mutations affecting the pivotal enzymatic activities, would be highly unlikely and probably incompatible with life. To date, thirteen TCA cycle-related genes have been described to be involved in the development of different cancers such as cutaneous and uterine leiomyomas, gastrointestinal tumors, gliomas, renal cancer, and especially PPGL. Thus, ~23% of PPGLs are found carrying mutations in genes encoding energy metabolism enzymes such as the succinate dehydrogenase (SDH) subunits (SDHx genes), fumarate hydratase or fumarase (FH), malate dehydrogenase 2 (MDH2), isocitrate dehydrogenases 1 (cytosolic), 2 and 3 (IDH1/2/3), glutamic-oxaloacetic transaminase 2 (GOT2) and solute carrier family 25 member 11 (SLC25A11) (Figure 1).
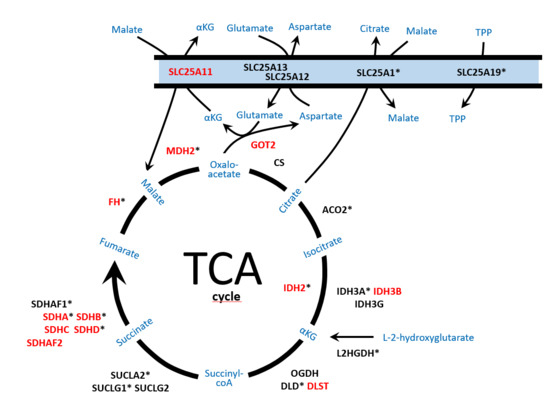
Figure 1.
Schematic representation of the enzymes and mitochondrial metabolic pathways, tricarboxylic acid (TCA) cycle, malate/aspartate shuttle, nicotinamide adenine dinucleotide (NADH) exchange and metabolite efflux from the mitochondria, involved in pheochromocytoma and paraganglioma (PPGL) development and/or neurodegenerative disorders. Enzymes reported as altered in PPGL are denoted in red capital letters. Enzymes involved in neurodegenerative disorders are denoted with an asterisk. SLC25A11: solute carrier family 25 member 11; SLC25A12/SLC25A13: carriers solute carrier family 25 members 12/13; SLC25A1: solute carrier family 25 member 1; SLC25A19: solute carrier family 25 member 19; GOT2: mitochondrial glutamic-oxaloacetic transaminase 2; FH: fumarate hydratase; MDH2: mitochondrial malate dehydrogenase; CS: citrate synthase; ACO2: mitochondrial aconitase; IDH2: isocitrate dehydrogenase 2; IDH3A/IDH3B/IDH3G: subunits of isocitrate dehydrogenase 3; L2HGDH: L-2-hydroxyglutarate dehydrogenase; OGDH/DLD/DLST: subunits of the αKG (alpha-ketoglutarate) dehydrogenase complex; SUCLA2/SUCLG1/SUCLG2: subunits of succinyl-CoA synthetase; SDHA/B/C/D: subunits of the succinate dehydrogenase complex; SDHAF1/SDHAF2: succinate dehydrogenase assembly factors; αKG: α-ketoglutarate; TPP: thiamine pyrophosphate.
PPGLs are neuroendocrine tumors derived from chromaffin cells of the adrenal medulla and from neural crest progenitors of extra-adrenal paraganglia. Sympathetic PPGLs, including PCC and thoracic-abdominal-pelvic (TAP) PGLs, are mostly catecholamine-producing tumors, whereas those derived from parasympathetic paraganglia, mainly located in the head and neck (H&N) region, are non-secreting tumors. Although PPGLs are predominantly benign and patients can be cured by surgical removal of these tumors, they present a significant morbidity and mortality due to the clinical aggressiveness of metastatic tumors (especially those carrying mutations in TCA cycle-related genes), for which therapeutic options remain scarce. To note, PPGL is considered a very rare disease with an incidence of 2–8 patients per million per year (1000–2000 new cases diagnosed worldwide every year). Therefore, the discovery of new susceptibility genes involved in hereditary predisposition to develop metastatic PPGLs, which is crucial for genetic counseling, guiding follow-up and developing targeted therapies, has only been possible through the joint efforts of many groups studying this extremely rare disease.
3. SDH Genes and PPGL
Germline loss-of-function mutations in SDHA, SDHB, SDHC, SDHD and SDHAF2 (together accounting for 20% of all PPGLs) cause the well-characterized familial PGL syndromes known as PGL5 [3], PGL4 [4], PGL3 [5], PGL1 [2] and PGL2 [6], respectively (Table 1). Additional solid tumors such as gastrointestinal stromal tumors (GISTs) [7], clear cell renal cell carcinomas (ccRCCs) [8] and pituitary adenomas (PAs) [9] have been associated, albeit rarely, with these familial PGL syndromes [10].

Table 1.
Summary of phenotypic and genetic features associated with the TCA cycle-related PPGL genes.
The SDH enzyme complex is a hetero-oligomer that comprises four structural subunits: two hydrophilic catalytic subunits, SDHA and SDHB, and two hydrophobic subunits that anchor the catalytic ones to the inner mitochondrial membrane, SDHC and SDHD. The fifth SDH gene associated with PPGL development, SDHAF2, encodes a cofactor responsible for flavination of SDHA. The SDH complex catalyzes the oxidative dehydrogenation of succinate to fumarate in the TCA cycle [11], and it is also functionally involved in the electron transport chain forming the mitochondrial complex II. Germline or somatic mutations in any of the SDH genes (SDHx) cause disassembly of the mitochondrial complex, with loss of SDH enzymatic activity and thus triggering the accumulation of its substrate, succinate. When succinate accumulates pathologically, it acts as a competitor of alpha-ketoglutarate (αKG) to broadly inhibit the activity of αKG-dependent dioxygenases, such as ten-eleven translocation (TET) DNA hydroxylases and Jumonji (JMJ) histone lysine demethylases (KDM) [12,13]. This causes a global hypermethylation with a characteristic CpG island methylation phenotype (CIMP) profile in the tumors, which leads to altered gene expression and contributes to tumorigenesis; this same mechanism was earlier observed in glioblastomas [14] and ccRCCs [15] carrying metabolic alterations such as IDH1/2 and FH/SDHB mutations, respectively. Apart from the aforementioned CIMP profile, the accumulation of succinate competitively inhibits the family of prolyl hydroxylase domain-containing proteins (PHD1-3), leading to hypoxia-inducible factor 1 α (HIF-1α) stabilization under normoxic conditions, and contributing to activation of the pseudohypoxic pathway [16,17]. More recently, it was reported that the succinate-mediated inhibition of two αKG-dependent dioxygenases, histone lysine demethylases KDM4A and KDM4B, leads to suppression of homologous recombination [18]. Moreover, the accumulation of succinate also causes downregulation of the enzyme responsible for the conversion of norepinephrine to epinephrine, thus inducing the characteristic noradrenergic phenotype of SDHx tumors [13]. All these processes orchestrated by succinate (and fumarate) accumulation (both referred to as oncometabolites) have been proposed to be involved in tumorigenesis (Figure 2) and/or in the particular phenotype of PPGLs carrying TCA cycle-related mutations. This will be discussed in more detail below.
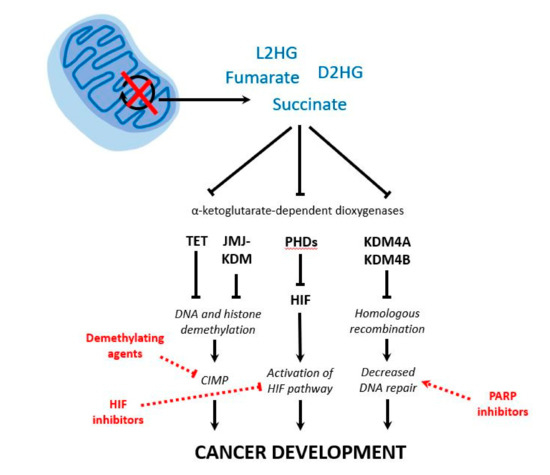
Figure 2.
Schematic representation of the consequences of tricarboxylic acid (TCA) cycle disruption in pheochromocytomas and paragangliomas (PPGL). Upon disruption of the activity of pivotal TCA cycle enzymes, there is an accumulation of metabolites (i.e., succinate, fumarate, D2HG and L2HG). Their efflux from the mitochondria to the cytosol and their subsequent competition with α-ketoglutarate lead to the inhibition of α-ketoglutarate-dependent dioxygenases involved in DNA and histone demethylation, regulation of HIF, and homologous recombination. As a result, different mechanisms are proposed as the cause of tumorigenesis in PPGL: aberrant global hypermethylation (CIMP), activation of the HIF pathway and decreased DNA repair. Finally, different therapeutic options may target each altered pathway: demethylating agents, HIF inhibitors, and poly-(ADP-ribose)-polymerase (PARP) inhibitors, respectively. D2HG: D-2-hydroxyglutarate; L2HG: L-2-hydroxyglutarate; TET: ten-eleven translocation DNA hydroxylase; JMJ: Jumonji; KDM: histone lysine demethylase; PHDs: prolyl hydroxylase domain-containing proteins; HIF: hypoxia-inducible factor; CIMP: CpG island methylation phenotype.
3.1. SDHD
Baysal and colleagues described in 2000 the first gene responsible for hereditary PGL [2]. SDHD, referred to at that time as PGL1 (the gene responsible for hereditary PGL type 1), had been mapped before to chromosome band 11q23, and was identified by direct sequencing of the best candidate gene contained in a subsequently narrowed critical interval [19]. The initial link with head and neck (H&N) PGLs, highly vascular tumors mainly arising in the main sensor of blood oxygenation (the carotid body), was rapidly extended to PCCs and TAP PGLs [20,21,22]. Although the original reports proposed that maternal imprinting accounted for the inheritance pattern observed in SDHD-related pedigrees, recent studies found that the “almost exclusive” paternal transmission of the disease can be explained by a somatic genetic mechanism targeting both the SDHD locus and a paternally imprinted gene on 11p15.5 [23]. Regardless the mechanism involved, the preferential paternal transmission of the disease may lead to generation skipping, making genetic counselling challenging. Moreover, though it can be considered as a rare scenario, development of PPGL may occur after maternal transmission of an SDHD mutation [24,25,26]. The estimated risk of PPGL in SDHD mutation carriers (excluding probands) at age 60 years is 43.2% [11]. Regarding the clinical presentation of SDHD mutations carriers, they primarily develop (multiple) H&N PGLs (84% of cases), although up to 22% also develop TAP PGLs and 12–24% develop PCCs (mainly unilateral); 3–10% of carriers develop metastases [27,28,29]. GISTs, PAs and, more rarely, ccRCCs can develop in SDHD mutation carriers [30]. It seems that the type of mutation, either impairing or not SDHD stability, influences the mutation-associated phenotype [27].
3.2. SDHB
One year after the finding of the first SDHD germline mutations in patients with PGL, mutations in SDHB, the gene encoding the SDH iron-sulfur subunit, were described as the genetic cause of hereditary PGL type 4 [4]. Soon, the presence of either point mutations or gross deletions affecting SDHB far surpassed the prevalence of SDHD mutations in patients with PPGL. Overall, mutations in SDHB and SDHD account for the great majority (over 60%) of all SDHx-related PGL patients, and SDHB itself accounts for approximately 10% of all PPGLs [31]. Unlike SDHD-associated clinical manifestations (i.e., multiple H&N PGLs), SDHB mutation carriers are usually diagnosed with single tumors that can arise in different locations (i.e., PCC, TAP PGL and H&N PGL) [32]. Paradoxically, mutations in SDHB show one of the lowest penetrances amongst the SDHx genes (13–21% at the age of 50 years) [11,31,33,34,35], but patients carrying germline SDHB mutations present a higher risk of malignancy (~50%) than other SDHx mutation carriers [11,32,36,37]. This latter observation favors the recommendation of prioritizing genetic testing of this gene in patients affected with PPGL [38]. In fact, the presence of mutations in SDHB is the best biomarker of poor prognosis and malignancy in PGL syndromes [36,39]. In addition, the absence of SDHB immunostaining in tumor cells, probably caused by altered assembly or SDH complex stability, is a reliable identifier of PPGLs caused not only by SDHB mutations, but also by any other SDHx mutation [40]. Although there is no explanation yet, it has been reported that male SDHB mutation carriers are at higher risk of disease than females [31]. Amongst the five SDHx genes, SDHB is the one with the most hereditary extra-paraganglial manifestations, such as ccRCCs [8,41], oncocytomas [42,43], GISTs [7], and PAs [44].
3.3. SDHC
The SDHC subunit anchors, along with SDHD, the SDH complex to the mitochondrial inner membrane. Though SDHC was the second SDHx gene identified as a cause of hereditary PGL (PGL type 3) [5], the frequency of patients carrying mutations in this gene is much lower than SDHB- and SDHD-related PPGLs, accounting for less than 1% of the patients. SDHC mutations result primarily in benign and non-functional H&N PGLs, but they have also been identified in patients with sympathetic PGLs [45,46]. Although no somatic point mutation affecting SDHC has been reported to date in PPGL, postzygotic epimutations in the gene promoter region have been identified in patients with PGLs [47], Carney triad (GIST, pulmonary chondroma and PGLs) [48], and Carney-Stratakis syndrome (PGL and GIST) [49]. This specific molecular mechanism of inactivation of the SDHC gene was first described in GISTs [50], and has been found in patients developing more than one PGL and/or having syndromic features resembling a hereditary case, as occurs with endothelial PAS domain protein 1 (EPAS1, also known as HIF2A) and H3F3A postzygotic somatic mutations. To identify these “genetically hidden” SDHx cases, a negative immunohistochemistry for SDHB appears to be essential. Although until now there are few patients reported, it seems that in general SDHC mutation carriers have low risk of developing GISTs and PAs [11].
3.4. SDHA
Paradoxically, mutations affecting the gene encoding one of the two major catalytic subunits of the SDH complex were described subsequently to mutations affecting the other members of the complex. Homozygous germline mutations of SDHA in patients with Leigh syndrome have been known since 1995 [51], but the involvement of this gene in the development of PPGL was elusive until 2010 [3]. This can be explained because, although SDHA variants are significantly enriched in PPGL cases, several SDHA alleles show a high prevalence amongst the normal population (i.e., 0.1–1% in gnomAD), and were therefore not considered as causative mutations in PPGL patients [52]. Mutations in SDHA have the lowest penetrance of all major PGL predisposition genes (10% at age 70 years) [53], and therefore most SDHA mutation carriers will not manifest the disease. Familial PPGL related to mutations in SDHA shows a prevalence of 3%, especially in patients with PGL, although it is not rare to find mutations in cases developing PCC [54]. The presence of metastasis in SDHA mutation carriers is 12%, and extraparaganglial manifestations such as PAs, GIST and ccRCC have also been described [53,54,55].
3.5. SDHAF2
Only two different mutations in SDHAF2, encoding one of the known SDH complex assembly factors, have been described to date in families with H&N PGLs [6,56,57], and one of them (i.e., p.Gly78Arg) exhibits a founder effect in the Dutch population [58]. As occurs with SDHD mutation carriers, carriers of SDHAF2 mutations show an exclusively paternal transmission of the disease, maybe because both genes are located on the same chromosome and may follow the same route to tumorigenesis [59]. Patients carrying mutations in SDHAF2 develop PGLs only in the H&N region, and frequently (74% of mutation carriers) in multiple locations [60,61]. Though the SDHAF1 gene has been found mutated in SDH-defective infantile leukoencephalopathy, no mutations have been reported in PPGL to date [62].
4. Other TCA Cycle-Related Genes
4.1. FH
Fumarate hydratase (FH) catalyzes the reversible hydration of fumarate to malate in the TCA cycle. Deficiency in FH activity leads to the accumulation of the oncometabolite fumarate and to the subsequent inhibition of multiple αKG-dependent enzymes, which drives critical epigenetic changes and signaling pathway activation. Thus, overabundance of fumarate, as occurs with succinate, leads to stabilization of HIF-1α, deregulation of DNA/histone methylation, increase of glutaminolysis and glycolysis, and production of reactive oxygen species (something not found in SDH-mutant PPGLs) [63]. All these altered phenomena could promote carcinogenesis by stimulating proliferation and cell survival. Inactivating germline mutations affecting the FH gene are the cause of hereditary leiomyomatosis and renal cell carcinoma (HLRCC) [64], but, as far as we know, no PPGL has been reported in families with HLRCC. In 2013, a germline mutation in FH was found in a patient with PCC by whole exome sequencing (WES) applied to a tumor displaying transcriptional and methylation (CIMP profile) similarities to SDH-mutant tumors [13]. Subsequently, the study of two large series of patients allowed the identification of additional patients carrying inactivating germline FH mutations [65,66]. The prevalence of alterations in the gene is about 1% in PPGL patients, and a metastatic phenotype and presence of multiple tumors located in the adrenal gland or in TAP paraganglia are the clinical characteristics of PPGLs associated with FH mutations. In addition, fumarate causes a non-enzymatic covalent modification of cysteine residues in proteins, S-(2-succinyl) cysteine (2SC) [67], that can be detected by immunohistochemistry and has been proposed as a biomarker for FH-associated neoplasia. This protein modification, called succination, is different from succinylation, another post-translational modification that will be discussed later in this review. The extent of succination and the implications it may have for the function of targeted proteins is poorly studied, but it seems that this process is more likely to lead to inactivation of critical enzymes [68,69].
4.2. MDH2
MDH2 encodes the enzyme malate dehydrogenase 2, which is essential for the reversible oxidation of malate to oxaloacetate in the TCA cycle. This tumor suppressor gene was first reported mutated, with an incomplete penetrance, in a single family with multiple malignant PGLs [70]. Very recently, additional variants have been reported, accounting for <1% of the patients, and their involvement in noradrenergic PPGLs with malignant behavior has been suggested [71]. Tumors carrying MDH2 mutations showed no accumulation of malate, and though knockdown of MDH2 in HeLa cells triggered the accumulation of both malate and fumarate, the connection between mutations in this gene and tumorigenesis is not clear. Subsequent to the finding of MDH2 mutations in PPGL, biallelic mutations of the gene were found as the cause of severe encephalopathy in pediatric patients [72], reinforcing the pathological role of alterations in this particular gene.
4.3. IDH Genes
Isocitrate dehydrogenases IDH1 (cytoplasmic) and IDH2 (mitochondrial) catalyze the oxidative decarboxylation of isocitrate to αKG. Recurrent, and mutually exclusive, mutations in the genes IDH1 (involving R132) and IDH2 (involving R172), result in neomorphic production of the oncometabolite D-2-hydroxyglutarate (D2HG) that ultimately causes the characteristic CIMP profile previously mentioned for succinate and fumarate accumulation [73]. Somatic mutations in the IDH1 gene (p.R132C), frequently found in central nervous system tumors [74], have been rarely identified in PGLs (i.e., three benign tumors in TAP or H&N locations) [49,75,76]. On the other hand, only one somatic IDH2 mutation has been found in a patient with a single H&N PGL by metabolome-guided genomic analysis [77]. Moreover, we recently found a germline truncating mutation affecting IDH3B in a patient with a single H&N PGL showing an altered αKG/isocitrate ratio and a CIMP-like profile [49]. Homozygous loss-of-function mutations in IDH3B have been found in families with retinitis pigmentosa, a hereditary neurodegeneration of rod and cone photoreceptors in the retina [78]. IDH3 is a heterotetramer, with two catalytic subunits encoded by IDH3A and two regulatory subunits encoded by IDH3B and IDH3G, which catalyzes the irreversible conversion of isocitrate to αKG in the TCA cycle. Although the finding of an IDH3B loss-of-function variant in a neuroendocrine tumor such as PGL suggests a causative role for this gene in the disease, and also for the other two genes encoding the remaining subunits of the tetramer, further studies are needed to definitively confirm this association.
4.4. SLC25A11 and GOT2
Another WES study applied to a tumor exhibiting an SDHx-like molecular phenotype (i.e., pseudohypoxic and CIMP profiles) in the absence of SDHx or FH mutations, identified a germline mutation in the SLC25A11 gene, which encodes the mitochondrial α-KG/malate carrier [79]. Five additional patients, most of them developing metastatic TAP PGLs, were found in this study carrying SLC25A11 mutations. This gene is not directly involved in the TCA cycle, but it participates in the exchange between two major intermediates of the cycle, which suggests that inactivation of other genes causing alterations in mitochondrial homeostasis can be responsible for PPGL development as well. Moreover, a single gain-of-function mutation in the GOT2 gene, encoding the mitochondrial glutamic-oxaloacetic transaminase and also involved in stimulating the malate/aspartate shuttle, was recently reported in a patient with multiple metastatic PGL [49], further reinforcing the link between dysfunction of proteins involved in the exchange of metabolites between the mitochondria and the cytoplasm and PPGL.
4.5. New TCA Cycle-Related Genes Involved in PPGL Development
Recently, a recurrent germline variant affecting DLST was found in PPGL patients with multiple tumors [80]. DLST (dihydrolipoamide S-succinyltransferase) is one of the three components (the E2 component) of the 2-oxoglutarate dehydrogenase (OGDH) complex that catalyzes the overall conversion of αKG to succinyl-CoA and CO2. Accumulation of L-2-hydroxyglutarate (L2HG) was found both in DLST-mutated tumors and in DLST-knockout (KO) cells transfected with the mutated protein. Surprisingly, and despite the mentioned accumulation, DLST-mutated tumors did not exhibit a CIMP profile, but they showed methylation and expression profiles similar to those observed for EPAS1-mutated PPGLs, suggesting a link between DLST disruption and pseudohypoxia. Moreover, a high HIF3A expression and a positive DLST immunostaining exclusively found in tumors carrying TCA cycle mutations or EPAS1 mutations further supported this pseudohypoxic link.
5. Metabolic Remodeling not Associated with TCA-Cycle Alterations
Apart from the mentioned TCA cycle alterations, metabolic reprograming of cancer cells can be achieved in PPGLs by other molecular mechanisms. Mutations in EPAS1, as well as the stabilization of HIF-1α occurring in VHL- and PHD-mutated PPGLs, trigger a pseudohypoxic switch of metabolism from mitochondrial respiration to glycolysis irrespective of oxygen levels. On the other hand, MYC deregulation caused by mutations in MAX may increase, in cooperation with HIF-2α, glucose uptake and glycolysis. Moreover, activating alterations of the phosphatidylinositol 3-kinase (PI3K)/AKT serine-threonine kinase (Akt)/mammalian target of rapamycin (mTOR) pathway (by loss-of-function mutations in NF1 and TMEM127, or gain-of-function mutations in RET, FGFR1 and HRAS) can also increase glycolysis through the transcription of glycolytic enzymes [63].
6. Inborn TCA Cycle Alterations: Neurodegenerative Disorders versus Cancer
The relevance of hereditary alterations in TCA cycle-related genes to the etiology of severe mitochondrial disorders is well known. Thus, autosomal-recessive mutations in almost any of the genes encoding the main enzymes of the TCA cycle lead to different forms of encephalopathies (Figure 1). This is the case for recessive germline mutations in SUCLG1 [81] and SUCLA2 [82]. Moreover, ACO2 alterations cause infantile cerebellar-retinal degeneration [83] and severe optic atrophy and spastic paraplegia [84]; IDH3A mutations lead to severe encephalopathy in infancy [85]; mutations in DLD cause severe encephalopathy and hyperlactatemia with neonatal onset [86]; homozygous IDH2 mutations provoke developmental delay, epilepsy, hypotonia, cardiomyopathy, and dysmorphic features [87]; SDHAF1 mutations lead to infantile leukoencephalopathy [62]; SDHA alteration is a well-known cause of Leigh syndrome, cardiomyopathy and leukodystrophy [51]; recessive SDHB mutations lead to hypotonia and leukodystrophy [88] and SDHD mutations cause encephalomyopathy [89]; FH alterations cause progressive encephalopathy in early childhood [90]; and MDH2 mutations provoke early-onset severe encephalopathy [72]. In addition, homozygous mutations in SLC25A1, encoding a mitochondrial citrate carrier, cause combined D-2- and L-2-hydroxyglutaric aciduria, severe neonatal epileptic encephalopathy, absence of developmental progress, and often early death [91], and mutations in SLC25A19, encoding a mitochondrial transporter of a TCA cycle cofactor (thiamine pyrophosphate), cause encephalopathy and progressive polyneuropathy [92]. Finally, L2HGDH alterations cause macrocephaly, developmental delay, epilepsy, and cerebellar ataxia [93].
Considering that complete abrogation of the TCA cycle in cells is highly unlikely to be compatible with cell viability, how is it possible that homozygous germline mutations in TCA cycle-related genes exist? The first mammalian model lacking a protein of the TCA cycle was obtained in 1997. Johnson et al. generated viable DLD+/− mice, while the homozygous knockout animals (DLD−/−) died at early embryonic stages [94]. This also occurred with SDHD [95], FH [96], DLST [97], SDHB [13] and SUCLA2/SUCLG2 [98] knockout mice. Overall, these data suggest that complete lack of activity of TCA cycle enzymes appears to be deleterious during embryonic development. These observations imply that homozygous or compound heterozygous variants in TCA cycle-related genes associated with neurological disorders do not completely abolish the corresponding enzymatic activity and retain part of their functionality. However, the presence of homozygous or compound heterozygous SDHx mutations in patients with neurological disorders gives rise to a controversial situation, which is the absence of tumors in mutation carriers. In heterozygous mutation carriers, this can be either accounted for by the low penetrance of TCA cycle-related mutations (especially in the case of SDHA and SDHB variants) or by the aforementioned modest effect of the alterations found in these patients. In addition, though clinical presentation and survival may be variable [99], patients carrying homozygous or compound heterozygous germline mutations in TCA cycle genes are usually diagnosed at a young age and die within a few years of diagnosis before reaching adulthood [100], so tumor diagnosis is unlikely to occur in these patients. However, taking into account that patients with L-2-hydroxyglutaric aciduria show increased risk of brain tumors [101], the tumorigenic potential of recessive mutations in TCA cycle-related genes cannot be ruled out. Both PPGL patients carrying TCA cycle-related mutations and patients with encephalopathies associated with the presence of recessive mutations in the aforementioned genes may benefit from therapeutic approaches targeting the aberrant DNA/histone methylation and the DNA-repair pathway, such as the use of demethylating agents (5-aza-2′-deoxycytidine or decitabine, for instance) or PARP inhibitors, respectively.
7. TCA Cycle-Related Omics Profiling in PPGLs
7.1. Pseudohypoxic Transcriptional Profile
The major regulator of cellular response to limited levels of oxygen (hypoxia) is the heterodimeric (composed of alpha and beta subunits) basic helix-loop-helix transcription factor HIF-1α. Under low oxygen conditions, HIF-1α (and also HIF-2α) activates the transcription of many genes involved in the metabolic adaptation to hypoxia, including transporters for increased glucose import (favoring anaerobic growth by glycolysis) and genes encoding angiogenesis factors [102]. The regulation of HIF-1α activity involves oxygen-limited hydroxylation of prolyl residues carried out by PHD1-3 prolyl hydroxylases. This prolyl hydroxylation induces the binding of HIF-1α to the von Hippel–Lindau (VHL) protein-associated complex, which is in charge of targeting HIF-1α by ubiquitination for proteosomal degradation [103,104]. Prolyl hydroxylation of HIF-1α requires oxygen, iron, and αKG, and the reaction produces succinate. Under hypoxia, prolyl hydroxylation is inhibited, HIF-1α is not ubiquitinated and this leads to the transcription of HIF-responsive genes. However, cancer cells may reproduce the hypoxic gene expression signature regardless of the oxygen condition in a process known as pseudohypoxia. This process is the hallmark of the VHL syndrome, in which mutations of VHL lead to HIF-1α stabilization in normoxia, leading to the development of tumors (e.g., ccRCCs and PPGLs). The pseudohypoxic transcriptional profile associated with the presence of mutations in TCA cycle-related genes in PPGL was first described by Patricia L Dahia in 2005. In this pioneering study, it was shown that SDH mutations, and the subsequent accumulation of succinate, caused a pseudohypoxic transcription profile similar to the one originated by VHL dysfunction [16,63,105]. Chronic pseudohypoxic signaling could be a mitogenic tumor initiator in neuroendocrine cells, and therefore inappropriate HIF-1α or HIF-2α persistence due to loss of SDH function in PPGL could drive tumorigenesis [106,107]. To note, the only environmental risk factor described in PPGL is chronic hypoxia which, in populations living at high altitude, leads to an increased incidence of H&N PGLs [108,109,110,111]. However, though the accumulation of oncometabolites is undoubtedly linked to tumor development in PPGLs, the subsequent pseudohypoxic response is not the only downstream mechanism proposed to explain tumorigenesis caused by TCA cycle dysfunction. The identification and validation of HIF-2α as one of the main oncogenic drivers in PPGLs [112,113,114] provides a rationale for exploring direct inhibition of HIF-2α as a therapeutic target in metastatic patients [115]. Preclinical studies have shown that HIF-2α antagonists are capable of inhibiting tumor growth in several models of renal cancer, and clinical trials of the first-in-class HIF-2α antagonist PT2385 [(S)-3-((2,2-difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1H-inden-4-yl)oxy)-5-fluorobenzonitrile] have achieved promising results in ccRCC [116]. These findings set the stage for future trials focused on metastatic PPGL, a tumor type that, similar to ccRCC, harbors the pseudo-hypoxia molecular signature as its main molecular hallmark [117,118].
7.2. TCA-Cycle Mutations and CpG Island Methylator Phenotype (CIMP)
The association between the presence of SDHx mutations and CIMP in PPGL has been known since 2008 [119]. Geli et al. performed a quantitative evaluation of promoter methylation of a set of tumor suppressor genes (i.e., RASSF1A, RASSF5, CDKN2A, RARB, TNFRSF10D, CDH1, and APC), and found that five of seven tumors exhibiting a targeted CIMP profile were mutated in SDHB. Moreover, this particular CIMP profile (defined by the presence of methylation in at least three of the genes included in the analysis) was associated with metastatic behavior and extra-adrenal location (both clinical characteristics related to SDHx mutations). Later, it was published that the accumulation of fumarate and succinate, upon FH or SDHx mutations respectively, led to enzymatic inhibition of multiple α-KG-dependent dioxygenases and consequent alterations of genome-wide histone and DNA methylation, linking the TCA cycle mutations to tumorigenesis by their effect on the epigenome [12]. Interestingly, and like a snake that bites its tail, the methylation of the promoter of SDHC is a very well-known mechanism leading to CIMP in GIST [50] and PPGLs [47,49]. In 2013, two separate studies based on DNA methylation profiling of samples carrying SDHx mutations further explored the connection between metabolic disruption and altered epigenetic modifications [13,120]. These studies uncovered that SDH deficiency, and the subsequent accumulation of succinate, led to DNA hypermethylation in multiple tumor lineages (e.g., PPGLs and GISTs). These epigenomic changes were particularly severe in SDHB-mutated tumors, potentially explaining their malignancy. To note, one of these methylation-based studies also uncovered mutations in FH as a cause of hereditary PPGL [13], expanding the CIMP to another TCA cycle-related gene beyond the SDHx genes. In addition, a sporadic PPGL has been also described showing CIMP associated with the presence of a IDH1 mutation [49] but, as previously mentioned, tumors carrying DLST mutations do not exhibit the characteristic CIMP profile despite their accumulation of TCA metabolites [80]. To date, the presence of mutations in other genes involved in the epigenetic machinery in PPGLs exhibiting hypermethylation [121,122], and the finding of tumors with a CIMP profile not associated with mutations in any TCA cycle-related gene [49], further suggest that other genes and pathways may be involved in this particular phenotype. Additional studies are required to elucidate whether the accumulation of metabolites, and the subsequent CIMP, is the cause or a consequence of the tumorigenic process and the adverse outcome associated with some TCA cycle mutations in PPGL.
7.3. Metabolome-Guided Genetic Characterization of the TCA Cycle
Since a link between pathological accumulation of TCA cycle metabolites and tumorigenesis was established, the study of the PPGL-associated metabolome has been used in the characterization of tumors carrying common genetic alterations, and additionally, in the identification of new candidate genes [49], the interpretation of genetic variants, and the improvement of diagnostics [77,123,124]. Mass spectrometry-based measurements of succinate:fumarate ratios allow to distinguish between SDH-deficient PPGLs and tumors without dysfunction of the SDH complex [123]. This is true, except in the case of some H&N PGLs, which may exhibit higher fumarate levels and therefore lower succinate:fumarate ratios compared to those at adrenal or TAP locations. The absence of this particular metabolic signature in some H&N PGLs is probably due to their higher content of stromal cells that dilute the signal from the tumor cells. In addition, alterations in the metabolites’ ratios can be used to identify hidden alterations in other TCA cycle-related genes. Thus, a tumor with a gain-of-function GOT2 mutation exhibited a high succinate:fumarate ratio, probably due to the accumulation of αKG and its subsequent conversion to succinate [49]. Moreover, high fumarate:malate ratios have been observed in PPGLs carrying FH mutations, high absolute values of D2HG can be detected in PPGLs carrying IDH1/2 mutations [49,77], and accumulation of L2HG was found both in DLST-mutated PPGLs and in DLST-KO cells transfected with the mutated protein [80].
In addition, hypermethylation (and the subsequent downregulation) of genes involved in the biosynthesis (PNMT, DRD2 and SULT1A1), transport (SLC6A2), and secretion (NPY) of catecholamines found in SDH- and FH-mutant PPGLs [13], may contribute to the immature catecholamine phenotypic features of PGLs carrying mutations in TCA cycle-related genes [124]. Thus, the reduced PNMT expression reported in SDH-mutant tumors has been proposed to cause the predominant secretion of noradrenaline or dopamine observed in these tumors.
8. Link Between Defective TCA Cycle and DNA Repair
On the whole, mutations affecting TCA cycle-related genes are associated with metastatic PPGLs for which curative chances, if any, are very limited. For that reason, the recently reported link between accumulation of TCA cycle oncometabolites and homologous recombination paves the way for new therapeutic approaches to the treatment of metastatic PPGL. Moreover, FH enzymatic activity is required for the cellular DNA damage response to double-strand breaks, and it is known that it is involved in the non-homologous end joining repair pathway [125,126]. Thus, in this unexpected connection between metabolism and DNA repair, the excess of fumarate and succinate not only would inhibit αKG-dependent dioxygenase activities, specifically the lysine demethylases KDM4A and KDM4B, but also would suppress the homologous recombination pathway [18]. This blockage would avoid the maintenance of genomic integrity and would make cells vulnerable to synthetic-lethal targeting with poly [adenosine diphosphate (ADP)]-ribose polymerase (PARP) inhibitors. Recent findings have revealed that the expression of mitochondrial complex I core subunits were upregulated in PPGLs with a pseudohypoxic profile. This augmented complex I activity increases intracellular nicotinamide adenine dinucleotide (NAD+) levels, which serves as an important cofactor to support the PARP DNA repair pathway. Thus, pseudohypoxic PPGLs would present more efficient DNA repair, resulting in potential chemoresistance.Interestingly, a combined treatment with a PARP inhibitor and temozolomide improved cytotoxicity in vitro, reducing tumor proliferation and metastatic lesions with prolonged overall survival in mice with SDHB-KO allografts [127].
9. Defective TCA Cycle Metabolism, Succinylation of Histones and Transcriptional Responses
In addition to the mentioned effects that the accumulation of TCA cycle metabolites has on overall DNA methylation, there are other less known implications of disruption of the cycle. Lysine succinylation is a known post-translational protein modification [128] that when occurring in histones may affect chromosome structure and function [129]. It was recently demonstrated that a fraction of the OGDH complex localizes in the nucleus and binds to lysine acetyltransferase 2A (KAT2A) in the promoter regions of genes [130], and this nuclear translocation of the complex depends on a nuclear localization sequence in DLST. Preventing the OGDH complex from entering the nucleus avoids nuclear generation of succinyl-CoA and the subsequent KAT2A-dependent H3 succinylation, reducing gene expression and inhibiting tumor cell proliferation and tumor growth. These results are consistent with previous observations linking nucleosome succinylation with enhanced in vitro transcription. These interesting findings not only might explain the tumorigenic potential of DLST, as part of the OGDH complex, but also open a new avenue of research into the connection of metabolism and cancer. A subsequent study also demonstrated a potential role of chromatin succinylation in modulating gene expression using an inducible cell culture model of SDH loss, which results in accumulation of succinyl-CoA [131]. This study also demonstrated that defective TCA cycle metabolism results in a DNA repair defect. Chromatin succinylation may thus represent a mechanism by which metabolism modulates both genome-wide transcription and DNA repair activities.
Apart from the aforementioned effect on the succinylation of histones, inhibition of the OGDH complex also reduces lysine succinylation of cytosolic and mitochondrial proteins altering rates of enzymes and pathways, especially mitochondrial metabolic pathways [132,133]. Moreover, ablation of specific enzymes of the TCA cycle affects the availability of succinyl CoA and global enzymatic and non-enzymatic succinylation patterns [134], providing a novel mechanism in which mitochondrial intermediates act as sensors to regulate metabolism.
10. Can We Open New Therapeutic Avenues for TCA Cycle-Altered PPGLs?
Apart from the known approaches to treating malignant PPGLs [135,136], we will discuss here novel therapeutic strategies that should be pursued for tumors carrying alterations in the TCA cycle. Though overall PPGLs present a low degree of chromosome instability, about 2–3% of tumors show chromothripsis involving chromosome arm 1p, where SDHB is located [129]. Chromothripsis phenomenon is a form of genome instability, associated with poor prognosis [137,138], in which one or a few chromosomes are affected by an alternating copy number profile with both loss and retention of heterozygosity [139,140]. One of the mechanisms involved in the initiation of chromothripsis is telomere dysfunction that leads to attrition of chromosome ends [141]. Subsequently, cancer cells showing this phenomenon become stabilized in order to avoid additional chromosomal aberrations that would be incompatible with cell survival. A well-known telomere stabilization mechanism involves the presence of high levels of TERT mRNA, mainly through amplification, promoter point mutations, methylation or rearrangements. Interestingly, increased TERT expression and telomere length has been observed in chromothripsis-positive ependymomas and glioblastomas [142], and in high-stage neuroblastomas [143] among other cancers, suggesting that telomere maintenance pathways may represent therapeutic targets in chromothripsis-positive tumors. With regard to PPGL, high to moderate TERT expression has been observed in 18–51% of patients, which are often metastatic cases [75,144,145], and in an important proportion of patients associated with deficiency of SDHx [146]. Moreover, the presence of somatic ATRX mutations, found preferentially in SDHB/FH-mutated tumors, and the associated alternative lengthening of telomeres have been described as an independent risk factor for metastatic PPGL [75,146,147]. It is tempting to speculate that metastatic PPGLs related to mutations in other TCA cycle genes that also exhibit telomere dysfunction, could benefit from the multiple telomerase-targeting therapeutic strategies that have been pursued in the past two decades (reviewed in [148]).
Another important avenue of potential treatments for metastatic PPGLs focuses on investigating whether a specific driver mutation could be associated with the tumor’s immune profile, and therefore with the potential efficacy of immunotherapy. In this regard, one of the main problems related to the potential lack of response is immune evasion, in which the tumor microenvironment (TME) plays a pivotal role [149]. The TME can be influenced by different conditions, but there are two in particular that gain importance when considering PPGLs with TCA-mutated genes: the hypoxic state, and an aberrant production of metabolites. Whether the PPGL immune subtypes are genotype-specific, or if there is an enrichment of any immune subtype among SDHB-mutated tumors or among those related to TCA genes with a higher metastatic risk is still to be addressed. Regardless these upcoming knowledge, it seems clear that a highly heterogeneous disease such as PPGL will benefit from a personalize treatment based on the specific genetic background, as well as from a deep characterization of the tumor microenvironment.
11. Conclusions
More than 20% of PPGLs are found carrying mutations in genes encoding TCA cycle metabolic enzymes. These mutations cause the disruption of the cycle, and the subsequent accumulation of “oncometabolites” lead to overall pathological changes in gene expression (i.e., by DNA methylation and post-translational protein modification), metabolic adaptation of cells to hypoxia, and DNA repair processes. Unraveling the pathological mechanisms associated with the presence of different PPGL mutations could pave the way to new personalized therapeutic approaches.
Author Contributions
Conceptualisation, A.C. and M.R.; Original draft preparation, A.C., L.R., B.C. and M.R.; Writing—review and editing, A.C., L.R., B.C. and M.R.; Supervision, A.C. and M.R.
Funding
This work was supported by the Instituto de Salud Carlos III (ISCIII), through the “Acción Estratégica en Salud” (AES) (projects PI18/00454 and PI17/01796, to A.C. and M.R., respectively, cofounded by the European Regional Development Fund (ERDF)), and the Paradifference Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Warburg, O. The metabolism of carcinoma cells 1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; Van Der Mey, A.; Taschner, P.E.M.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Brière, J.J.; Libé, R.; Vescovo, L.; Rivière, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Bénit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.M.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene Mutations in the Succinate Dehydrogenase Subunit SDHB Cause Susceptibility to Familial Pheochromocytoma and to Familial Paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. Familial Gastrointestinal Stromal Tumors and Germ-Line Mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Buchta, M.; McWhinney, S.R.; Virta, S.K.; Peçzkowska, M.; Morrison, C.D.; Lehtonen, R.; Januszewicz, A.; Järvinen, H.; Juhola, M.; et al. Early-Onset Renal Cell Carcinoma as a Novel Extraparaganglial Component of SDHB-Associated Heritable Paraganglioma. Am. J. Hum. Genet. 2004, 74, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C.A.; Rustin, P.; Nesterova, M.; De La Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J. Clin. Endocrinol. Metab. 2012, 97, 357–366. [Google Scholar] [CrossRef]
- Mannelli, M.; Canu, L.; Ercolino, T.; Rapizzi, E.; Martinelli, S.; Parenti, G.; De Filpo, G.; Nesi, G. DIAGNOSIS of ENDOCRINE DISEASE: SDHx mutations: Beyond pheochromocytomas and paragangliomas. Eur. J. Endocrinol. 2018, 178, R11–R17. [Google Scholar] [CrossRef]
- Andrews, K.A.; Ascher, D.B.; Pires, D.E.V.; Barnes, D.R.; Vialard, L.; Casey, R.T.; Bradshaw, N.; Adlard, J.; Aylwin, S.; Brennan, P.; et al. Tumour risks and genotype–phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 2018, 55, 384–394. [Google Scholar]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Ricketts, C.; Killian, J.K.; Vocke, C.D.; Sourbier, C.; Yang, Y.; Merino, M.J.; Meltzer, P.S.; Linehan, W.M. Abstract 2660: A renal CpG island methylator phenotype (R-CIMP) in kidney tumors associated with germline mutations of FH and SDHB. Cancer Res. 2016, 76, 2660. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Baysal, B.E.; Van Schothorst, E.M.; Farr, J.E.; Grashof, P.; Myssiorek, D.; Rubinstein, W.S.; Taschner, P.; Cornelisse, C.J.; Devlin, B.; Devilee, P.; et al. Repositioning the hereditary paraganglioma critical region on chromosome band 11q23. Hum. Genet. 1999, 104, 219–225. [Google Scholar] [CrossRef]
- Astuti, D.; Douglas, F.; Lennard, T.W.J.; Aligianis, I.A.; Woodward, E.R.; Evans, D.G.R.; Eng, C.; Latif, F.; Maher, E.R. Germline SDHD mutation in familial phaeochromocytoma. Lancet 2001, 357, 1181–1182. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-Line Mutations in Nonsyndromic Pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Cascon, A.; Ruiz-Llorente, S.; Cebrian, A.; Telleria, D.; Rivero, J.C.; Diez, J.J.; Lopez-Ibarra, P.J.; Jaunsolo, M.A.; Benitez, J.; Robledo, M. Identification of novel SDHD mutations in patients with phaeochromocytoma and/or paraganglioma. Eur. J. Hum. Genet. 2002, 10, 457–461. [Google Scholar] [CrossRef]
- Hensen, E.F.; Jordanova, E.S.; Van Minderhout, I.J.H.M.; Hogendoorn, P.C.W.; Taschner, P.E.M.; Van Der Mey, A.G.L.; Devilee, P.; Cornelisse, C.J. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene 2004, 23, 4076–4083. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Mazzella, J.M.; Drui, D.; Amar, L.; Bertherat, J.; Coupier, I.; Delemer, B.; Guilhem, I.; Herman, P.; Kerlan, V.; et al. Risk assessment of maternally inherited SDHD paraganglioma and phaeochromocytoma. J. Med. Genet. 2017, 54, 100–103. [Google Scholar] [CrossRef]
- Pigny, P.; Vincent, A.; Bauters, C.C.; Bertrand, M.; De Montpreville, V.T.; Crepin, M.; Porchet, N.; Caron, P. Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J. Clin. Endocrinol. Metab. 2008, 93, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Yeap, P.M.; Tobias, E.S.; Mavraki, E.; Fletcher, A.; Bradshaw, N.; Freel, E.M.; Cooke, A.; Murday, V.A.; Davidson, H.R.; Perry, C.G.; et al. Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J. Clin. Endocrinol. Metab. 2011, 96, E2009–E2013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ricketts, C.J.; Forman, J.R.; Rattenberry, E.; Bradshaw, N.; Lalloo, F.; Izatt, L.; Cole, T.R.; Armstrong, R.; Ajith Kumar, V.K.; Morrison, P.J.; et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 2010, 31, 41–51. [Google Scholar] [CrossRef]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef]
- Cascón, A.; Pita, G.; Burnichon, N.; Landa, I.; López-Jiménez, E.; Montero-Conde, C.; Leskelä, S.; Leandro-García, L.J.; Letón, R.; Rodríguez-Antona, C.; et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J. Clin. Endocrinol. Metab. 2009, 94, 1701–1705. [Google Scholar] [CrossRef]
- Evenepoel, L.; Papathomas, T.G.; Krol, N.; Korpershoek, E.; De Krijger, R.R.; Persu, A.; Dinjens, W.N.M. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet. Med. 2015, 17, 610–620. [Google Scholar] [CrossRef]
- Jochmanova, I.; Wolf, K.I.; King, K.S.; Nambuba, J.; Wesley, R.; Martucci, V.; Raygada, M.; Adams, K.T.; Prodanov, T.; Fojo, A.T.; et al. SDHB-related pheochromocytoma and paraganglioma penetrance and genotype–phenotype correlations. J. Cancer Res. Clin. Oncol. 2017, 143, 1421–1435. [Google Scholar] [CrossRef]
- Niemeijer, N.D.; Rijken, J.A.; Eijkelenkamp, K.; Van Der Horst-Schrivers, A.N.A.; Kerstens, M.N.; Tops, C.M.J.; Van Berkel, A.; Timmers, H.J.L.M.; Kunst, H.P.M.; Leemans, C.R.; et al. The phenotype of SDHB germline mutation carriers: A nationwide study. Eur. J. Endocrinol. 2017, 177, 115–125. [Google Scholar] [CrossRef]
- Rijken, J.A.; Niemeijer, N.D.; Jonker, M.A.; Eijkelenkamp, K.; Jansen, J.C.; van Berkel, A.; Timmers, H.J.L.M.; Kunst, H.P.M.; Bisschop, P.H.L.T.; Kerstens, M.N.; et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin. Genet. 2018, 93, 60–66. [Google Scholar] [CrossRef]
- Schiavi, F.; Milne, R.L.; Anda, E.; Blay, P.; Castellano, M.; Opocher, G.; Robledo, M.; Cascón, A. Are we overestimating the penetrance of mutations in SDHB? Hum. Mutat. 2010, 31, 761–762. [Google Scholar] [CrossRef]
- Jafri, M.; Whitworth, J.; Rattenberry, E.; Vialard, L.; Kilby, G.; Kumar, A.V.; Izatt, L.; Lalloo, F.; Brennan, P.; Cook, J.; et al. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin. Endocrinol. 2013, 78, 898–906. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.-P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.-F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar]
- Dhir, M.; Li, W.; Hogg, M.E.; Bartlett, D.L.; Carty, S.E.; McCoy, K.L.; Challinor, S.M.; Yip, L. Clinical Predictors of Malignancy in Patients with Pheochromocytoma and Paraganglioma. Ann. Surg. Oncol. 2017, 24, 3624–3630. [Google Scholar] [CrossRef]
- Cascón, A.; López-Jiménez, E.; Landa, I.; Leskelä, S.; Leandro-García, L.J.; Maliszewska, A.; Letón, R.; Vega, L.D.L.; García-Barcina, M.J.; Sanabria, C.; et al. Rationalization of genetic testing in patients with apparently sporadic pheochromocytoma/paraganglioma. Horm. Metab. Res. 2009, 41, 672–675. [Google Scholar] [CrossRef]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef]
- Van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.; Sleddens, H.F.; Derkx, P.; Rivière, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Cascón, A.; Landa, Í.; López-Jiménez, E.; Díez-Hernández, A.; Buchta, M.; Montero-Conde, C.; Leskelä, S.; Leandro-García, L.J.; Letón, R.; Rodríguez-Antona, C.; et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J. Med. Genet. 2008, 45, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.; Douglas, F.; Perros, P.; Morgan, C.; Maher, E.R. SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam. Cancer 2009, 8, 257–260. [Google Scholar] [CrossRef]
- Dénes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, M.; Ercolino, T.; Giachè, V.; Simi, L.; Cirami, C.; Parenti, G. Genetic screening for pheochromocytoma: Should SDHC gene analysis be included? J. Med. Genet. 2007, 44, 586–587. [Google Scholar] [CrossRef][Green Version]
- Else, T.; Marvin, M.L.; Everett, J.N.; Gruber, S.B.; Arts, H.A.; Stoffel, E.M.; Auchus, R.J.; Raymond, V.M. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J. Clin. Endocrinol. Metab. 2014, 99, E1482–E1486. [Google Scholar] [CrossRef]
- Richter, S.; Klink, B.; Nacke, B.; De Cubas, A.A.; Mangelis, A.; Rapizzi, E.; Meinhardt, M.; Skondra, C.; Mannelli, M.; Robledo, M.; et al. Epigenetic mutation of the succinate dehydrogenase c promoter in a patient with two paragangliomas. J. Clin. Endocrinol. Metab. 2016, 101, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef]
- Remacha, L.; Comino-Mendez, I.; Richter, S.; Contreras, L.; Curras-Freixes, M.; Pita, G.; Leton, R.; Galarreta, A.; Torres-Perez, R.; Honrado, E.; et al. Targeted exome sequencing of Krebs cycle genes reveals candidate cancer–predisposing mutations in pheochromocytomas and paragangliomas. Clin. Cancer Res. 2017, 23, 6315–6325. [Google Scholar] [CrossRef]
- Killian, J.K.; Miettinen, M.; Walker, R.L.; Wang, Y.; Zhu, Y.J.; Waterfall, J.J.; Noyes, N.; Retnakumar, P.; Yang, Z.; Smith, W.I.; et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci. Transl. Med. 2014, 6, 268ra177. [Google Scholar] [CrossRef]
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar] [CrossRef]
- Maniam, P.; Zhou, K.; Lonergan, M.; Berg, J.N.; Goudie, D.R.; Newey, P.J. Pathogenicity and penetrance of germline SDHA variants in pheochromocytoma and paraganglioma (PPGL). J. Endocr. Soc. 2018, 2, 806–816. [Google Scholar] [CrossRef]
- Van Der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.J.; Corssmit, E.P.M.; Dinjens, W.N.; Van De Horst-Schrivers, A.N.; Jansen, J.C.; De Jong, M.M.; Kunst, H.P.M.; Kusters, B.; et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: A nationwide study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445. [Google Scholar] [CrossRef]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef]
- Jha, A.; de Luna, K.; Balili, C.A.; Millo, C.; Paraiso, C.A.; Ling, A.; Gonzales, M.K.; Viana, B.; Alrezk, R.; Adams, K.T.; et al. Clinical, Diagnostic, and Treatment Characteristics of SDHA-Related Metastatic Pheochromocytoma and Paraganglioma. Front. Oncol. 2019, 9, 53. [Google Scholar] [CrossRef]
- Bayley, J.P.; Kunst, H.P.M.; Cascon, A.; Sampietro, M.L.; Gaal, J.; Korpershoek, E.; Hinojar-Gutierrez, A.; Timmers, H.J.L.M.; Hoefsloot, L.H.; Hermsen, M.A.; et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010, 11, 366–372. [Google Scholar] [CrossRef]
- Piccini, V.; Rapizzi, E.; Bacca, A.; Di Trapani, G.; Pulli, R.; Giachè, V.; Zampetti, B.; Lucci-Cordisco, E.; Canu, L.; Corsini, E.; et al. Head and neck paragangliomas: Genetic spectrum and clinical variability in 79 consecutive patients. Endocr. Relat. Cancer 2012, 19, 149–155. [Google Scholar] [CrossRef]
- Hensen, E.F.; van Duinen, N.; Jansen, J.C.; Corssmit, E.P.M.; Tops, C.M.J.; Romijn, J.A.; Vriends, A.H.J.T.; van der Mey, A.G.L.; Cornelisse, C.J.; Devilee, P.; et al. High prevalence of founder mutations of the succinate dehydrogenase genes in The Netherlands. Clin. Genet. 2012, 81, 284–288. [Google Scholar] [CrossRef]
- Hensen, E.F.; Siemers, M.D.; Jansen, J.C.; Corssmit, E.P.M.; Romijn, J.A.; Tops, C.M.J.; Van Der Mey, A.G.L.; Devilee, P.; Cornelisse, C.J.; Bayley, J.P.; et al. Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin. Endocrinol. 2011, 75, 650–655. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Kunst, H.P.M.; Rutten, M.H.; De Mönnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.L.M.; Marres, H.A.M.; Jansen, J.C.; Kremer, H.; Bayley, J.P.; Cremers, C.W.R.J. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer the multiple leiomyoma consortium. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascón, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef]
- Clark, G.R.; Sciacovelli, M.; Gaude, E.; Walsh, D.M.; Kirby, G.; Simpson, M.A.; Trembath, R.C.; Berg, J.N.; Woodward, E.R.; Kinning, E.; et al. Germline FH mutations presenting with pheochromocytoma. J. Clin. Endocrinol. Metab. 2014, 99, E2046–E2050. [Google Scholar] [CrossRef]
- Bardella, C.; El-Bahrawy, M.; Frizzell, N.; Adam, J.; Ternette, N.; Hatipoglu, E.; Howarth, K.; O’Flaherty, L.; Roberts, I.; Turner, G.; et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 2011, 225, 4–11. [Google Scholar] [CrossRef]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of Mitochondrial Aconitase by Succination in Fumarate Hydratase Deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef]
- Frizzell, N.; Lima, M.; Baynes, J.W. Succination of proteins in diabetes. Free Radic. Res. 2011, 45, 101–109. [Google Scholar] [CrossRef]
- Cascon, A.; Comino-Mendez, I.; Curras-Freixes, M.; de Cubas, A.A.; Contreras, L.; Richter, S.; Peitzsch, M.; Mancikova, V.; Inglada-Perez, L.; Perez-Barrios, A.; et al. Whole-Exome Sequencing Identifies MDH2 as a New Familial Paraganglioma Gene. JNCI J. Natl. Cancer Inst. 2015, 107, djv053. [Google Scholar] [CrossRef]
- Calsina, B.; Currás-Freixes, M.; Buffet, A.; Pons, T.; Contreras, L.; Letón, R.; Comino-Méndez, I.; Remacha, L.; Calatayud, M.; Obispo, B.; et al. Role of MDH2 pathogenic variant in pheochromocytoma and paraganglioma patients. Genet. Med. 2018, 20, 1652–1662. [Google Scholar] [CrossRef]
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; François, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.M.; Vasnier, C.; et al. Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Gaal, J.; Burnichon, N.; Korpershoek, E.; Roncelin, I.; Bertherat, J.; Plouin, P.F.; De Krijger, R.R.; Gimenez-Roqueplo, A.P.; Dinjens, W.N.M. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2010, 95, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Gieldon, L.; Pang, Y.; Peitzsch, M.; Huynh, T.; Leton, R.; Viana, B.; Ercolino, T.; Mangelis, A.; Rapizzi, E.; et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet. Med. 2018, 21, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Dange, M.; McGee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [CrossRef]
- Buffet, A.; Morin, A.; Castro-Vega, L.J.; Habarou, F.; Lussey-Lepoutre, C.; Letouze, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef]
- Remacha, L.; Pirman, D.; Mahoney, C.E.; Coloma, J.; Calsina, B.; Currás-Freixes, M.; Letón, R.; Torres-Pérez, R.; Richter, S.; Pita, G.; et al. Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. Am. J. Hum. Genet. 2019, 104, 651–664. [Google Scholar] [CrossRef]
- Carrozzo, R.; Verrigni, D.; Rasmussen, M.; de Coo, R.; Amartino, H.; Bianchi, M.; Buhas, D.; Mesli, S.; Naess, K.; Born, A.P.; et al. Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: Phenotype and genotype correlations in 71 patients. J. Inherit. Metab. Dis. 2016, 39, 243–252. [Google Scholar] [CrossRef]
- Carrozzo, R.; Dionisi-Vici, C.; Steuerwald, U.; Lucioli, S.; Deodato, F.; Di Giandomenico, S.; Bertini, E.; Franke, B.; Kluijtmans, L.A.J.; Meschini, M.C.; et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007, 130, 862–874. [Google Scholar] [CrossRef]
- Spiegel, R.; Pines, O.; Ta-Shma, A.; Burak, E.; Shaag, A.; Halvardson, J.; Edvardson, S.; Mahajna, M.; Zenvirt, S.; Saada, A.; et al. Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2. Am. J. Hum. Genet. 2012, 90, 518–523. [Google Scholar] [CrossRef]
- Bouwkamp, C.G.; Afawi, Z.; Fattal-Valevski, A.; Krabbendam, I.E.; Rivetti, S.; Masalha, R.; Quadri, M.; Breedveld, G.J.; Mandel, H.; Tailakh, M.A.; et al. ACO2 homozygous missense mutation associated with complicated hereditary spastic paraplegia. Neurol. Genet. 2018, 4, e223. [Google Scholar] [CrossRef]
- Fattal-Valevski, A.; Eliyahu, H.; Fraenkel, N.I.D.; Elmaliach, G.; Hausman-Kedem, M.; Shaag, A.; Mandel, D.; Pines, O.; Elpeleg, O. Homozygous mutation, p. Pro304His, in IDH3A, encoding isocitrate dehydrogenase subunit is associated with severe encephalopathy in infancy. Neurogenetics 2017, 18, 57–61. [Google Scholar] [CrossRef]
- Odièvre, M.-H.; Chretien, D.; Munnich, A.; Robinson, B.H.; Dumoulin, R.; Masmoudi, S.; Kadhom, N.; Rötig, A.; Rustin, P.; Bonnefont, J.-P. A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of α-ketoglutarate dehydrogenase deficiency. Hum. Mutat. 2005, 25, 323–324. [Google Scholar] [CrossRef]
- Kranendijk, M.; Struys, E.A.; Van Schaftingen, E.; Gibson, K.M.; Kanhai, W.A.; Van Der Knaap, M.S.; Amiel, J.; Buist, N.R.; Das, A.M.; De Klerk, J.B.; et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 2010, 330, 336. [Google Scholar] [CrossRef]
- Alston, C.L.; Davison, J.E.; Meloni, F.; van der Westhuizen, F.H.; He, L.; Hornig-Do, H.T.; Peet, A.C.; Gissen, P.; Goffrini, P.; Ferrero, I.; et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2012, 49, 569–577. [Google Scholar] [CrossRef]
- Jackson, C.B.; Nuoffer, J.M.; Hahn, D.; Prokisch, H.; Haberberger, B.; Gautschi, M.; Häberli, A.; Gallati, S.; Schaller, A. Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2014, 51, 170–175. [Google Scholar] [CrossRef]
- Bourgeron, T.; Chretien, D.; Poggi-Bach, J.; Doonan, S.; Rabier, D.; Letouzé, P.; Munnich, A.; Rötig, A.; Landrieu, P.; Rustin, P. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J. Clin. Investig. 1994, 93, 2514–2518. [Google Scholar] [CrossRef]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; Van Dooren, S.J.M.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Shaag, A.; Edvardson, S.; Mandel, H.; Stepensky, P.; Shalev, S.A.; Horovitz, Y.; Pines, O.; Elpeleg, O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 2009, 66, 419–424. [Google Scholar] [CrossRef]
- Rzem, R.; Veiga-da-Cunha, M.; Noel, G.; Goffette, S.; Nassogne, M.-C.; Tabarki, B.; Scholler, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef]
- Johnson, M.T.; Yang, H.-S.; Magnuson, T.; Patel, M.S. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality (gene targeting embryonic lethal mutation embryonic metabolism). Dev. Boil. 1997, 94, 14512–14517. [Google Scholar]
- Piruat, J.I.; Pintado, C.O.; Ortega-Saenz, P.; Roche, M.; Lopez-Barneo, J. The Mitochondrial SDHD Gene Is Required for Early Embryogenesis, and Its Partial Deficiency Results in Persistent Carotid Body Glomus Cell Activation with Full Responsiveness to Hypoxia. Mol. Cell. Biol. 2004, 24, 10933–10940. [Google Scholar] [CrossRef]
- Pollard, P.J.; Spencer-Dene, B.; Shukla, D.; Howarth, K.; Nye, E.; El-Bahrawy, M.; Deheragoda, M.; Joannou, M.; McDonald, S.; Martin, A.; et al. Targeted Inactivation of Fh1 Causes Proliferative Renal Cyst Development and Activation of the Hypoxia Pathway. Cancer Cell 2007, 11, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, Q.; Ho, D.J.; Starkov, A.A.; Wille, E.J.; Xu, H.; Chen, H.L.; Zhang, S.; Stack, C.M.; Calingasan, N.Y.; et al. Mice deficient in dihydrolipoyl succinyl transferase show increased vulnerability to mitochondrial toxins. Neurobiol. Dis. 2009, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kacso, G.; Ravasz, D.; Doczi, J.; Nemeth, B.; Madgar, O.; Saada, A.; Ilin, P.; Miller, C.; Ostergaard, E.; Iordanov, I.; et al. Two transgenic mouse models for-subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations. Biochem. J. 2016, 473, 3463–3485. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Hargreaves, I.P.; Duncan, A.J.; Taanman, J.W.; Heales, S.J.; Land, J.M.; Bitner-Glindzicz, M.; Leonard, J.V.; Rahman, S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 2006, 89, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. 2010, 18, 1160–1165. [Google Scholar] [CrossRef]
- Aghili, M.; Zahedi, F.; Rafiee, E. Hydroxyglutaric aciduria and malignant brain tumor: A case report and literature review. J. Neurooncol. 2009, 91, 233–236. [Google Scholar] [CrossRef]
- Maher, L.J., III; Smith, E.H.; Rueter, E.M.; Becker, N.A.; Bida, J.P.; Nelson-Holte, M.; Palomo, J.I.P.; García-Flores, P.; López-Barneo, J.; van Deursen, J. Mouse Models of Human Familial Paraganglioma. In Pheochromocytoma-A New View of the Old Problem; Intech: London, UK, 2011. [Google Scholar]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.M.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIf1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Her, Y.F.; Maher, L.J. Succinate dehydrogenase loss in familial paraganglioma: Biochemistry, genetics, and epigenetics. Int. J. Endocrinol. 2015, 2015, 296167. [Google Scholar] [CrossRef] [PubMed]
- Jochmanová, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Waguespack, S.G.; Rich, T.; Grubbs, E.; Ying, A.K.; Perrier, N.D.; Ayala-Ramirez, M.; Jimenez, C. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2010, 95, 2023–2037. [Google Scholar] [CrossRef]
- Astrom, K.; Cohen, J.E.; Willett-Brozick, J.E.; Aston, C.E.; Baysal, B.E. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: Evidence for an oxygen-sensing defect. Hum. Genet. 2003, 113, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Cerecer-Gil, N.Y.; Figuera, L.E.; Llamas, F.J.; Lara, M.; Escamilla, J.G.; Ramos, R.; Estrada, G.; Hussain, A.K.; Gaal, J.; Korpershoek, E.; et al. Mutation of SDHB is a cause of hypoxia-related high-altitude paraganglioma. Clin. Cancer Res. 2010, 16, 4148–4154. [Google Scholar] [CrossRef]
- Opotowsky, A.R.; Moko, L.E.; Ginns, J.; Rosenbaum, M.; Greutmann, M.; Aboulhosn, J.; Hageman, A.; Kim, Y.; Deng, L.X.; Grewal, J.; et al. Pheochromocytoma and paraganglioma in cyanotic congenital heart disease. J. Clin. Endocrinol. Metab. 2015, 100, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A Gain-of-Function Mutations in Paraganglioma with Polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef]
- Comino-Méndez, I.; de Cubas, A.A.; Bernal, C.; Álvarez-Escolá, C.; Sánchez-Malo, C.; Ramírez-Tortosa, C.L.; Pedrinaci, S.; Rapizzi, E.; Ercolino, T.; Bernini, G.; et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum. Mol. Genet. 2013, 22, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Qin, Y.; Srikantan, S.; Morales, N.P.; Li, Q.; Deng, Y.; Kim, S.W.; Pereira, M.A.A.; Toledo, S.P.A.; Su, X.; et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2013, 20, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.; Jimenez, C. Recent advances in the management of malignant pheochromocytoma and paraganglioma: Focus on tyrosine kinase and hypoxia-inducible factor inhibitors. F1000Research 2018, 7, 1148. [Google Scholar] [CrossRef]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2a antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kaelin, W.G. Targeting HIF2 in Clear Cell Renal Cell Carcinoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 113–121. [Google Scholar] [CrossRef]
- Geli, J.; Kiss, N.; Karimi, M.; Lee, J.J.; Bäckdahl, M.; Ekström, T.J.; Larsson, C. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: Association to malignant behavior. Clin. Cancer Res. 2008, 14, 2551–2559. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Remacha, L.; Currás-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018, 20, 1644–1651. [Google Scholar] [CrossRef]
- Toledo, R.A.; Qin, Y.; Cheng, Z.M.; Gao, Q.; Iwata, S.; Silva, G.M.; Prasad, M.L.; Ocal, I.T.; Rao, S.; Aronin, N.; et al. Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas. Clin. Cancer Res. 2016, 22, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Peitzsch, M.; Rapizzi, E.; Lenders, J.W.; Qin, N.; De Cubas, A.A.; Schiavi, F.; Rao, J.U.; Beuschlein, F.; Quinkler, M.; et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 2014, 99, 3903–3911. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.M.; Robledo, M. Metabologenomics of phaeochromocytoma and paraganglioma: An integrated approach for personalised biochemical and genetic testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar] [PubMed]
- Leshets, M.; Silas, Y.B.H.; Lehming, N.; Pines, O. Fumarase: From the TCA Cycle to DNA Damage Response and Tumor Suppression. Front. Mol. Biosci. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Yogev, O.; Singer, E.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the dna damage response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NADþ/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J., III. Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.M.; Skinner, M.E.; et al. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Weinert, B.T.; Schölz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Lowery, A.J.; Walsh, S.; McDermott, E.W.; Prichard, R.S. Molecular and therapeutic advances in the diagnosis and management of malignant pheochromocytomas and paragangliomas. Oncologist 2013, 18, 391–407. [Google Scholar] [CrossRef]
- Jimenez, C. Treatment for patients with malignant pheochromocytomas and paragangliomas: A perspective from the hallmarks of cancer. Front. Endocrinol. 2018, 9, 227. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Van Sluis, P.; Valentijn, L.J.; Van Der Ploeg, I.; Hamdi, M.; Van Nes, J.; Westerman, B.A.; Van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.W.; Zapatka, M.; Stütz, A.M.; Zichner, T.; Weischenfeldt, J.; Jäger, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Wilson, R.K. Chromothripsis and human disease: Piecing together the shattering process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef]
- Ernst, A.; Jones, D.T.W.; Maass, K.K.; Rode, A.; Deeg, K.I.; Jebaraj, B.M.C.; Korshunov, A.; Hovestadt, V.; Tainsky, M.A.; Pajtler, K.W.; et al. Telomere dysfunction and chromothripsis. Int. J. Cancer 2016, 138, 2905–2914. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; Van Sluis, P.; Volckmann, R.; Van Noesel, M.M.; George, R.E.; Tytgat, G.A.M.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Dwight, T.; Flynn, A.; Amarasinghe, K.; Benn, D.E.; Lupat, R.; Li, J.; Cameron, D.L.; Hogg, A.; Balachander, S.; Candiloro, I.L.M.; et al. TERT structural rearrangements in metastatic pheochromocytomas. Endocr. Relat. Cancer 2018, 25, 1–9. [Google Scholar] [CrossRef]
- Liu, T.; Brown, T.C.; Juhlin, C.C.; Andreasson, A.; Wang, N.; Bäckdahl, M.; Healy, J.M.; Prasad, M.L.; Korah, R.; Carling, T.; et al. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr. Relat. Cancer 2014, 21, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Job, S.; Draskovic, I.; Burnichon, N.; Buffet, A.; Cros, J.; Lépine, C.; Venisse, A.; Robidel, E.; Verkarre, V.; Meatchi, T.; et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2019, 25, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Khare, S.; Wubbenhorst, B.; Desloover, D.; D’Andrea, K.; Merrill, S.; Cho, N.W.; Greenberg, R.A.; Else, T.; Montone, K.; et al. Whole exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6140. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Goldkorn, A. Telomere and telomerase therapeutics in cancer. Genes 2016, 7, 22. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).