Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
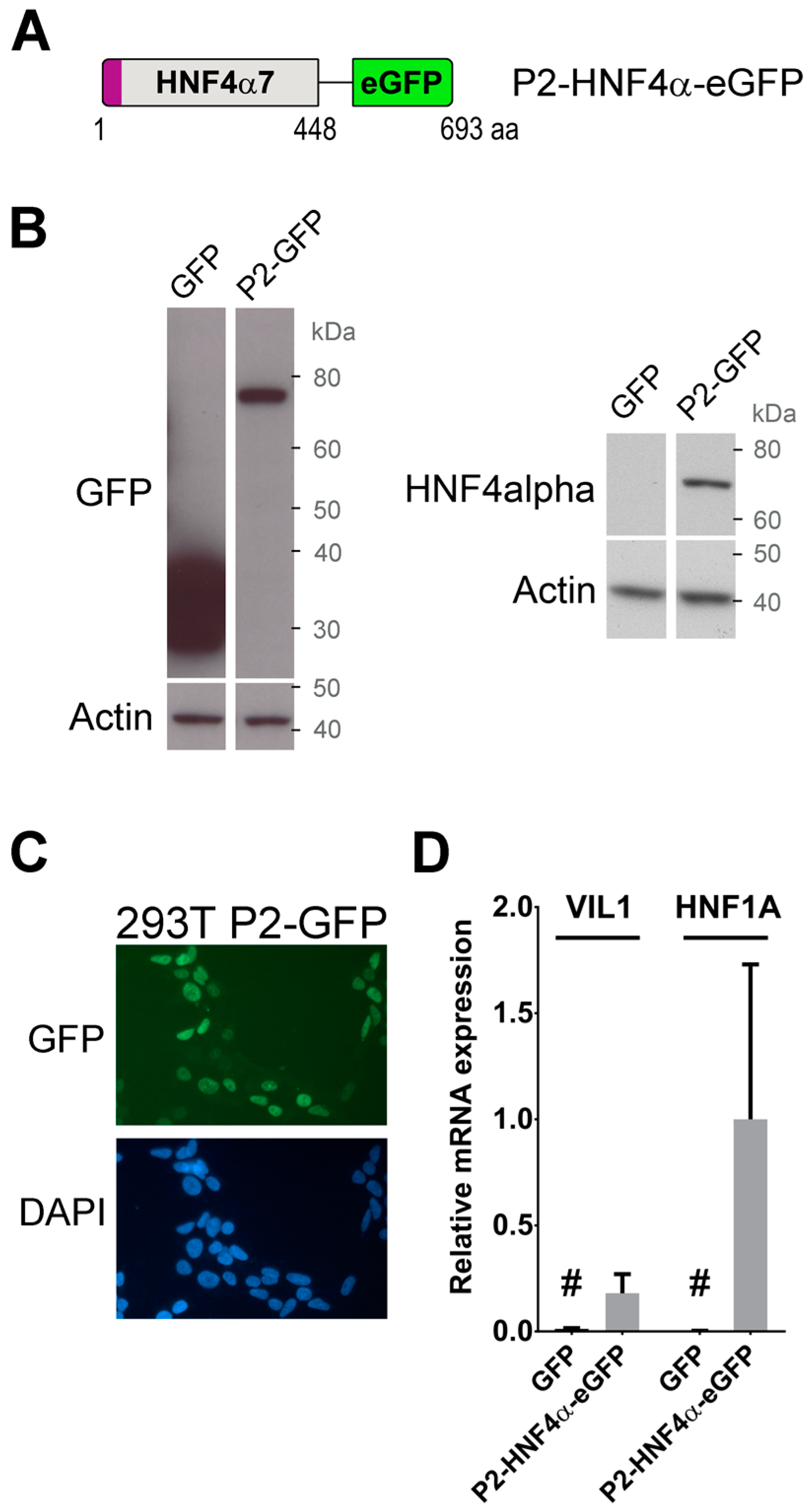
2.1. Generation of Cells Stably Expressing A Transcriptionally Functional P2-HNF4α Fusion Protein Construct
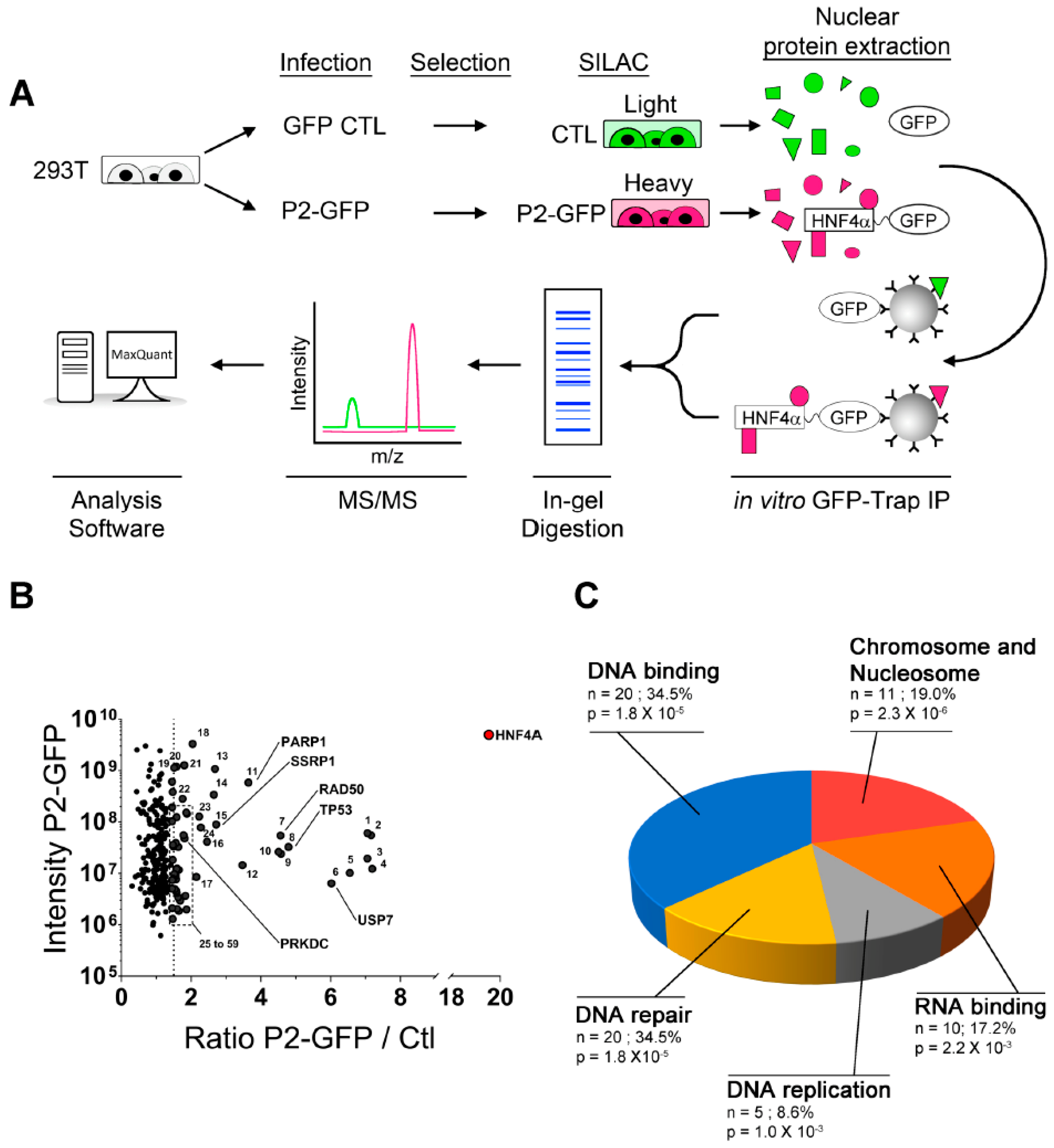
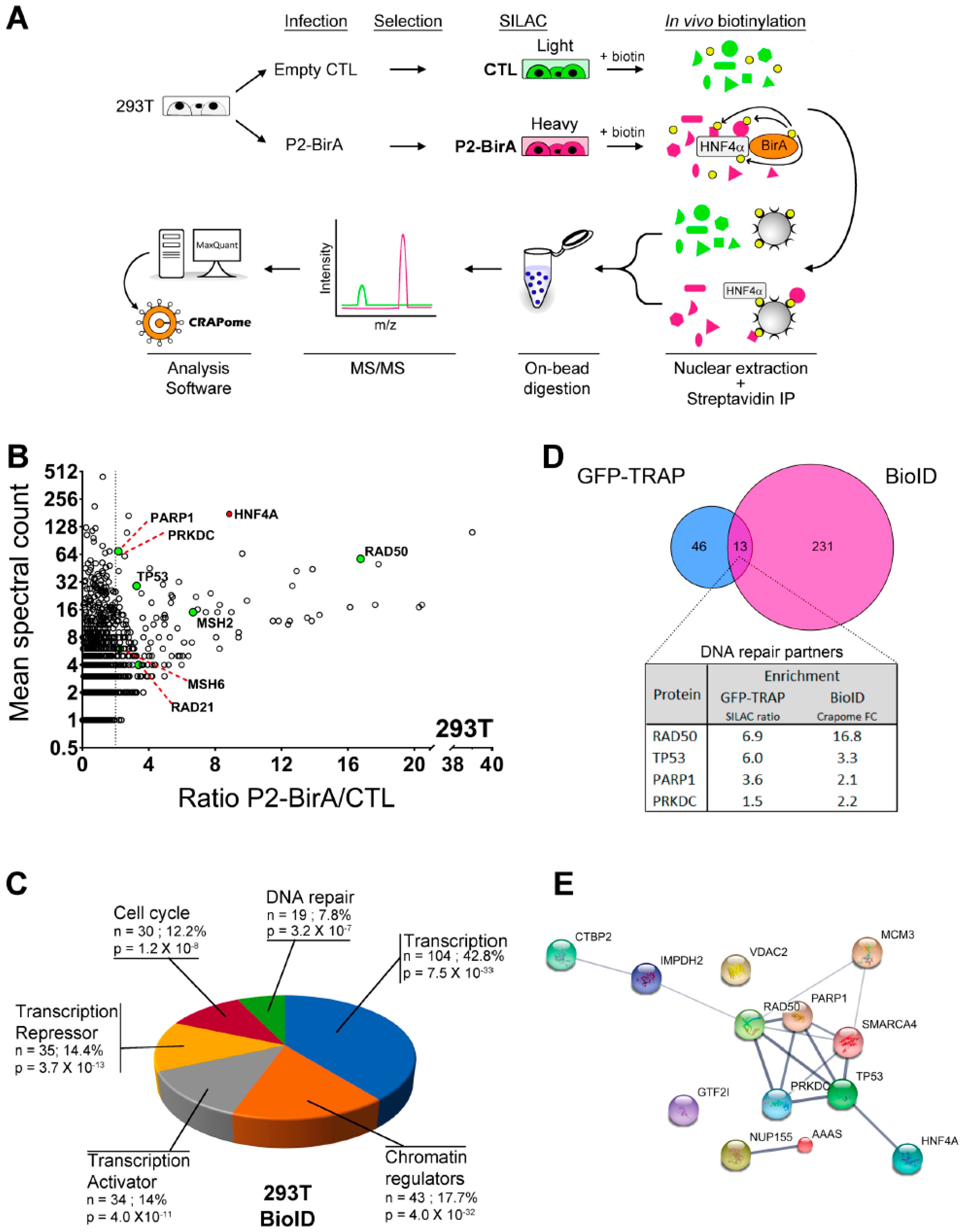
2.2. Novel P2-HNF4α Protein Interactomes Identified by Quantitative Proteomics in HEK293T Cells
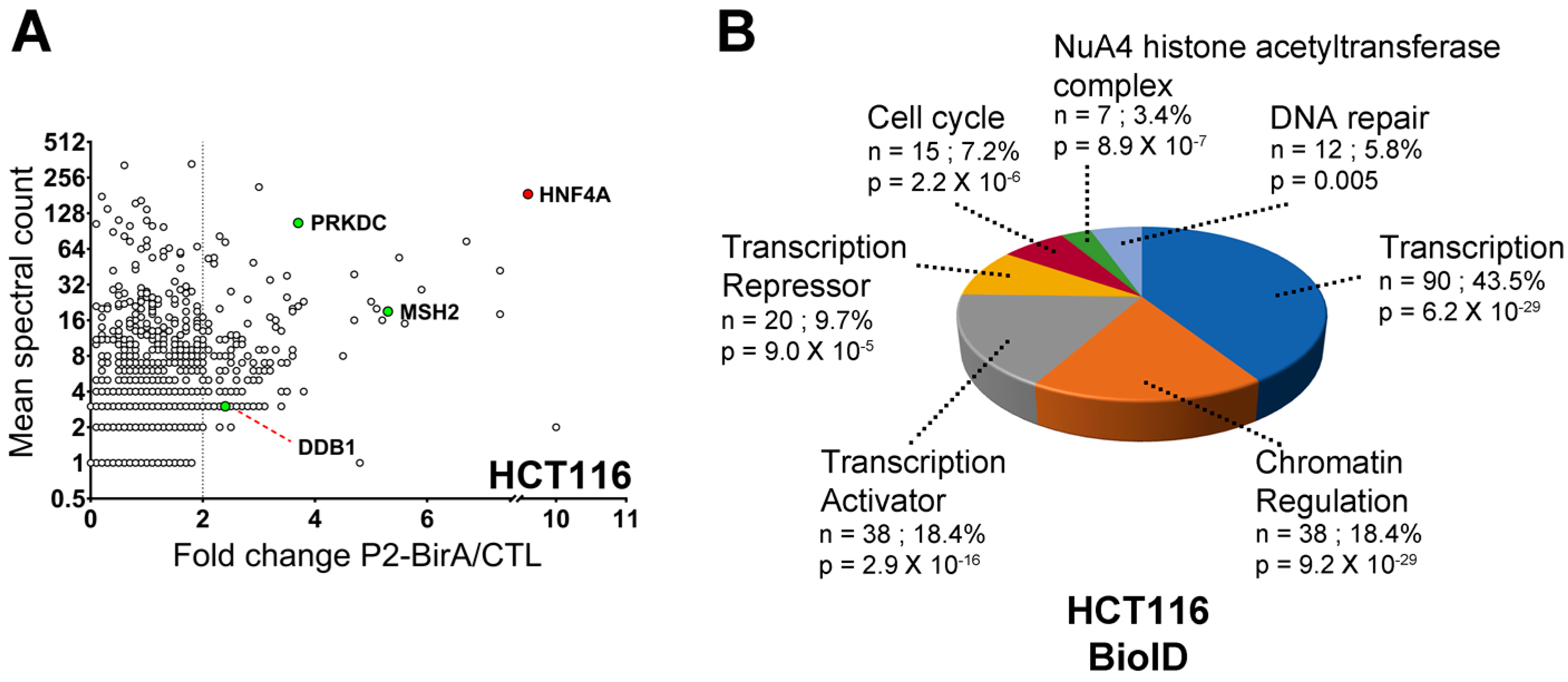
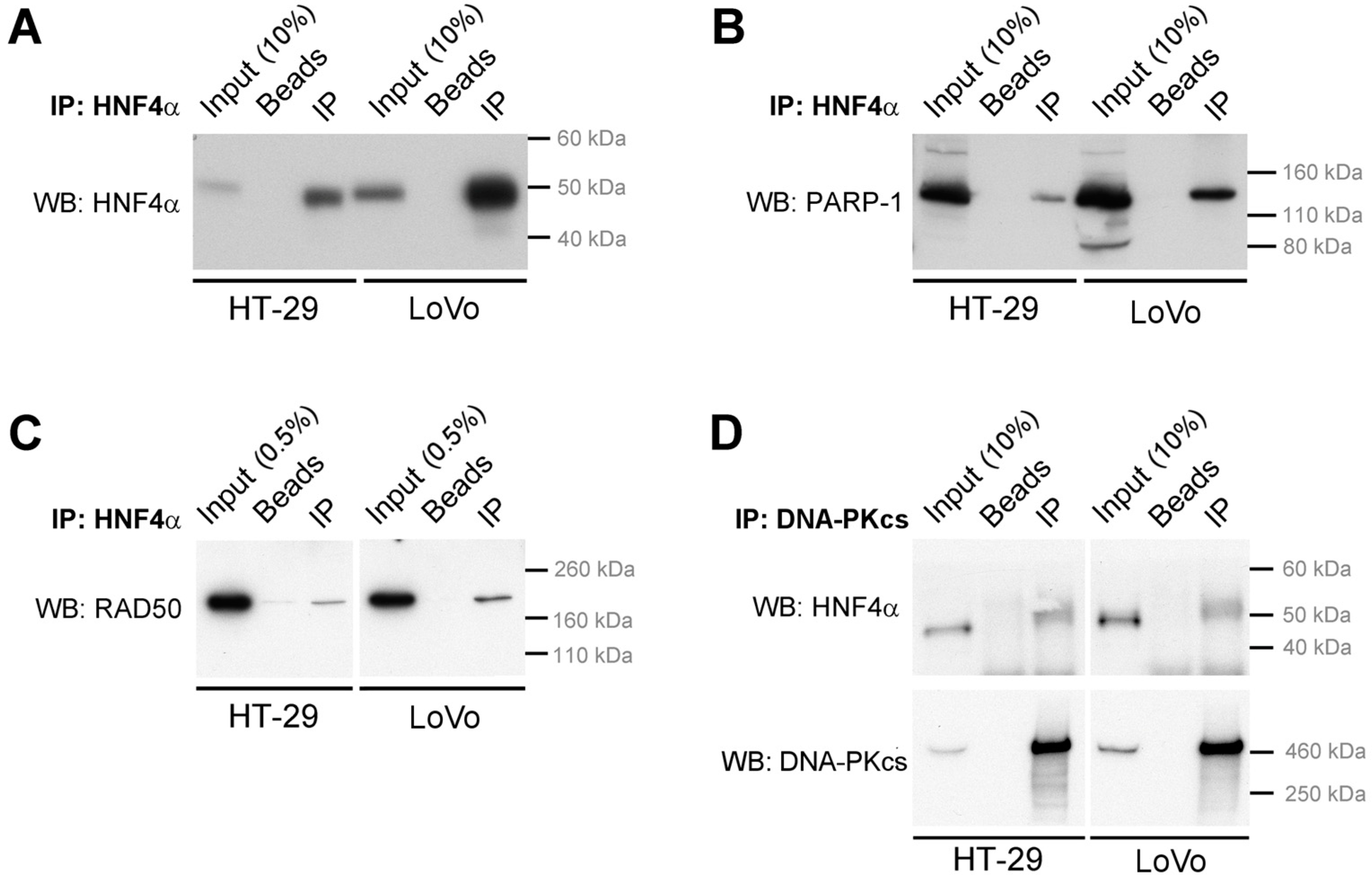
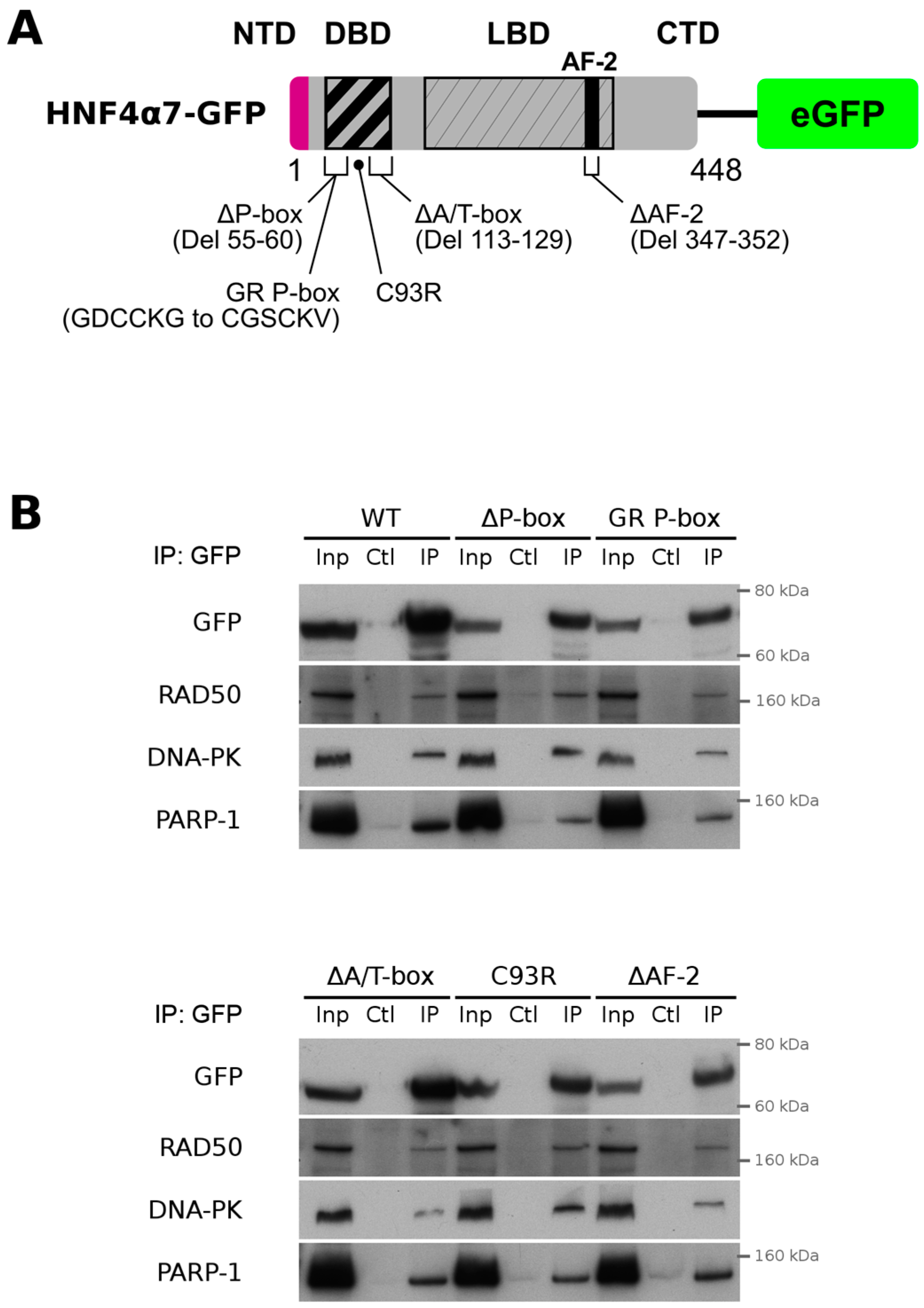
2.3. HNF4α Recruits DNA Damage Response Protein Partners in Colorectal Cancer Cells
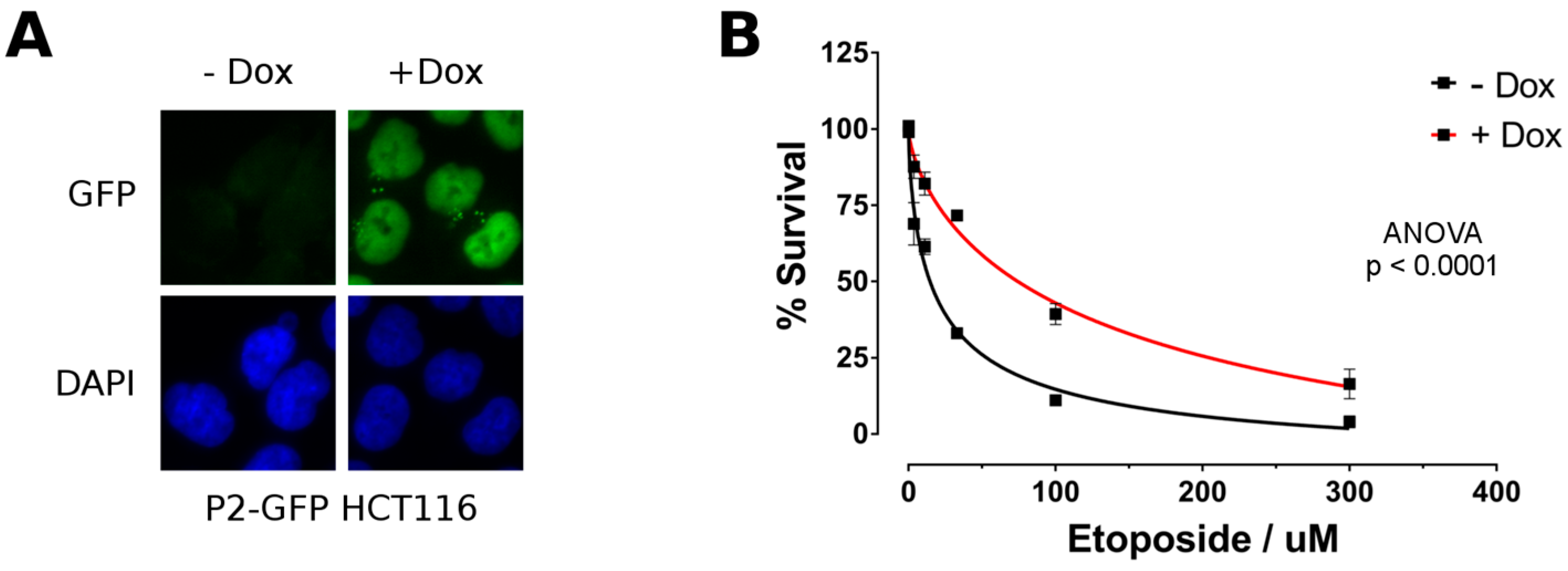
2.4. P2-GFP Decreases Sensitivity to Etoposide in HCT116 Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture and SILAC Labelling
4.2. Plasmid Construction and Generation of Stable Cell Lines
4.3. Quantitative PCR
4.4. Immunoprecipitations and Immunoblots
4.5. GFP-Trap Assay and In-Gel Digestion
4.6. BioID Assay
4.7. Etoposide Sensitivity Assay
4.8. Fluorescence Microscopy
4.9. Mass Spectrometry Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babeu, J.P.; Darsigny, M.; Lussier, C.R.; Boudreau, F. Hepatocyte nuclear factor 4alpha contributes to an intestinal epithelial phenotype in vitro and plays a partial role in mouse intestinal epithelium differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G124–G134. [Google Scholar] [CrossRef]
- Chen, W.S.; Manova, K.; Weinstein, D.C.; Duncan, S.A.; Plump, A.S.; Prezioso, V.R.; Bachvarova, R.F.; Darnell, J.E., Jr. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 1994, 8, 2466–2477. [Google Scholar] [CrossRef]
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Jiang, S.; Hotta, H.; Takano, K.; Iwanari, H.; Sumi, K.; Daigo, K.; Ohashi, R.; Sugai, M.; Ikegame, C.; et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J. Pathol. 2006, 208, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Sladek, F.M.; Zhong, W.M.; Lai, E.; Darnell, J.E., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4, 2353–2365. [Google Scholar] [CrossRef]
- Odom, D.T.; Zizlsperger, N.; Gordon, D.B.; Bell, G.W.; Rinaldi, N.J.; Murray, H.L.; Volkert, T.L.; Schreiber, J.; Rolfe, P.A.; Gifford, D.K.; et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004, 303, 1378–1381. [Google Scholar] [CrossRef]
- Cattin, A.L.; Le Beyec, J.; Barreau, F.; Saint-Just, S.; Houllier, A.; Gonzalez, F.J.; Robine, S.; Pincon-Raymond, M.; Cardot, P.; Lacasa, M.; et al. Hepatocyte nuclear factor 4alpha, a key factor for homeostasis, cell architecture, and barrier function of the adult intestinal epithelium. Mol. Cell. Biol. 2009, 29, 6294–6308. [Google Scholar] [CrossRef]
- Lussier, C.R.; Babeu, J.P.; Auclair, B.A.; Perreault, N.; Boudreau, F. Hepatocyte nuclear factor-4alpha promotes differentiation of intestinal epithelial cells in a coculture system. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G418–G428. [Google Scholar] [CrossRef] [PubMed]
- Gougelet, A.; Colnot, S. MicroRNA-feedback loop as a key modulator of liver tumorigenesis and inflammation. World J. Gastroenterol. 2013, 19, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, S.N.; Anjum, K.M.; Gui, L.X.; Zhu, S.S.; Liu, J.; Chen, J.K.; Liu, Q.F.; Ye, G.D.; Wang, W.J.; et al. A double-negative feedback loop between Wnt-beta-catenin signaling and HNF4alpha regulates epithelial-mesenchymal transition in hepatocellular carcinoma. J. Cell Sci 2013, 126, 5692–5703. [Google Scholar] [CrossRef] [PubMed]
- Darsigny, M.; Babeu, J.P.; Seidman, E.G.; Gendron, F.P.; Levy, E.; Carrier, J.; Perreault, N.; Boudreau, F. Hepatocyte nuclear factor-4alpha promotes gut neoplasia in mice and protects against the production of reactive oxygen species. Cancer Res. 2010, 70, 9423–9433. [Google Scholar] [CrossRef]
- Schwartz, B.; Algamas-Dimantov, A.; Hertz, R.; Nataf, J.; Kerman, A.; Peri, I.; Bar-Tana, J. Inhibition of colorectal cancer by targeting hepatocyte nuclear factor-4alpha. Int. J. Cancer 2009, 124, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Babeu, J.P.; Boudreau, F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J. Gastroenterol. 2014, 20, 22–30. [Google Scholar] [CrossRef]
- Babeu, J.P.; Jones, C.; Geha, S.; Carrier, J.C.; Boudreau, F. P1 promoter-driven HNF4alpha isoforms are specifically repressed by beta-catenin signaling in colorectal cancer cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Vuong, L.M.; Chellappa, K.; Dhahbi, J.M.; Deans, J.R.; Fang, B.; Bolotin, E.; Titova, N.V.; Hoverter, N.P.; Spindler, S.R.; Waterman, M.L.; et al. Differential Effects of Hepatocyte Nuclear Factor 4alpha Isoforms on Tumor Growth and T-Cell Factor 4/AP-1 Interactions in Human Colorectal Cancer Cells. Mol. Cell. Biol. 2015, 35, 3471–3490. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, K.; Deol, P.; Evans, J.R.; Vuong, L.M.; Chen, G.; Briancon, N.; Bolotin, E.; Lytle, C.; Nair, M.G.; Sladek, F.M. Opposing roles of nuclear receptor HNF4alpha isoforms in colitis and colitis-associated colon cancer. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Delia, D.; Mizutani, S.; Panigone, S.; Tagliabue, E.; Fontanella, E.; Asada, M.; Yamada, T.; Taya, Y.; Prudente, S.; Saviozzi, S.; et al. ATM protein and p53-serine 15 phosphorylation in ataxia-telangiectasia (AT) patients and at heterozygotes. Br. J. Cancer 2000, 82, 1938–1945. [Google Scholar] [CrossRef]
- Ou, Y.H.; Chung, P.H.; Sun, T.P.; Shieh, S.Y. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol. Biol. Cell 2005, 16, 1684–1695. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. p53: Master of life, death, and the epigenome. Genes Dev. 2017, 31, 955–956. [Google Scholar] [CrossRef] [PubMed]
- Malewicz, M.; Perlmann, T. Function of transcription factors at DNA lesions in DNA repair. Exp. Cell. Res. 2014, 329, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, J.; Mittal, S.; Wang, Y.; Korkmaz, K.S.; Adamson, E.; English, C.; Ohmichi, M.; McClelland, M.; Mercola, D. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol. Cell 2004, 16, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, K.; Jankova, L.; Schnabl, J.M.; Pan, S.; Brelivet, Y.; Fung, C.L.; Chan, C.; Dent, O.F.; Clarke, S.J.; Robertson, G.R.; et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform-specific loss of HNF4alpha in human colon cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, E.; Liao, H.; Ta, T.C.; Yang, C.; Hwang-Verslues, W.; Evans, J.R.; Jiang, T.; Sladek, F.M. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology 2010, 51, 642–653. [Google Scholar] [CrossRef]
- Verzi, M.P.; Shin, H.; San Roman, A.K.; Liu, X.S.; Shivdasani, R.A. Intestinal master transcription factor CDX2 controls chromatin access for partner transcription factor binding. Mol. Cell. Biol. 2013, 33, 281–292. [Google Scholar] [CrossRef]
- Lambert, E.; Babeu, J.-P.; Simoneau, J.; Levesque, D.; Jolibois, E.; Scott, M.; Boudreau, F.; Boisvert, F.-M. Human Hepatocyte Nuclear Factor 4a encodes isoforms with distinct transcriptional functions. bioRxiv 2019, 585604. [Google Scholar]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell. Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Boulon, S.; Lam, Y.W.; Urcia, R.; Boisvert, F.M.; Vandermoere, F.; Morrice, N.A.; Swift, S.; Rothbauer, U.; Leonhardt, H.; et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 2008, 183, 223–239. [Google Scholar] [CrossRef]
- Malewicz, M.; Kadkhodaei, B.; Kee, N.; Volakakis, N.; Hellman, U.; Viktorsson, K.; Leung, C.Y.; Chen, B.; Lewensohn, R.; van Gent, D.C.; et al. Essential role for DNA-PK-mediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev. 2011, 25, 2031–2040. [Google Scholar] [CrossRef]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef]
- Maeda, Y.; Seidel, S.D.; Wei, G.; Liu, X.; Sladek, F.M. Repression of hepatocyte nuclear factor 4alpha tumor suppressor p53: Involvement of the ligand-binding domain and histone deacetylase activity. Mol. Endocrinol. 2002, 16, 402–410. [Google Scholar] [CrossRef]
- Poletto, M.; Lirussi, L.; Wilson, D.M., 3rd; Tell, G. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol. Biol. Cell 2014, 25, 1641–1652. [Google Scholar] [CrossRef]
- Chen, Z.; Tran, M.; Tang, M.; Wang, W.; Gong, Z.; Chen, J. Proteomic Analysis Reveals a Novel Mutator S (MutS) Partner Involved in Mismatch Repair Pathway. Mol. Cell. Proteom. 2016, 15, 1299–1308. [Google Scholar] [CrossRef]
- Dubois, M.L.; Bastin, C.; Levesque, D.; Boisvert, F.M. Comprehensive Characterization of Minichromosome Maintenance Complex (MCM) Protein Interactions Using Affinity and Proximity Purifications Coupled to Mass Spectrometry. J. Proteome Res. 2016, 15, 2924–2934. [Google Scholar] [CrossRef]
- Asselin-Mullen, P.; Chauvin, A.; Dubois, M.L.; Drissi, R.; Levesque, D.; Boisvert, F.M. Protein interaction network of alternatively spliced NudCD1 isoforms. Sci. Rep. 2017, 7, 12987. [Google Scholar] [CrossRef]
- Boisvert, F.M.; Lam, Y.W.; Lamont, D.; Lamond, A.I. A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage. Mol. Cell. Proteom. 2010, 9, 457–470. [Google Scholar] [CrossRef]
- Drissi, R.; Dubois, M.L.; Douziech, M.; Boisvert, F.M. Quantitative Proteomics Reveals Dynamic Interactions of the Minichromosome Maintenance Complex (MCM) in the Cellular Response to Etoposide Induced DNA Damage. Mol. Cell. Proteom. 2015, 14, 2002–2013. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babeu, J.-P.; Wilson, S.D.; Lambert, É.; Lévesque, D.; Boisvert, F.-M.; Boudreau, F. Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells. Cancers 2019, 11, 626. https://doi.org/10.3390/cancers11050626
Babeu J-P, Wilson SD, Lambert É, Lévesque D, Boisvert F-M, Boudreau F. Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells. Cancers. 2019; 11(5):626. https://doi.org/10.3390/cancers11050626
Chicago/Turabian StyleBabeu, Jean-Philippe, Samuel D. Wilson, Élie Lambert, Dominique Lévesque, François-Michel Boisvert, and François Boudreau. 2019. "Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells" Cancers 11, no. 5: 626. https://doi.org/10.3390/cancers11050626
APA StyleBabeu, J.-P., Wilson, S. D., Lambert, É., Lévesque, D., Boisvert, F.-M., & Boudreau, F. (2019). Quantitative Proteomics Identifies DNA Repair as a Novel Biological Function for Hepatocyte Nuclear Factor 4α in Colorectal Cancer Cells. Cancers, 11(5), 626. https://doi.org/10.3390/cancers11050626