Repurposing the Electron Transfer Reactant Phenazine Methosulfate (PMS) for the Apoptotic Elimination of Malignant Melanoma Cells through Induction of Lethal Oxidative and Mitochondriotoxic Stress



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
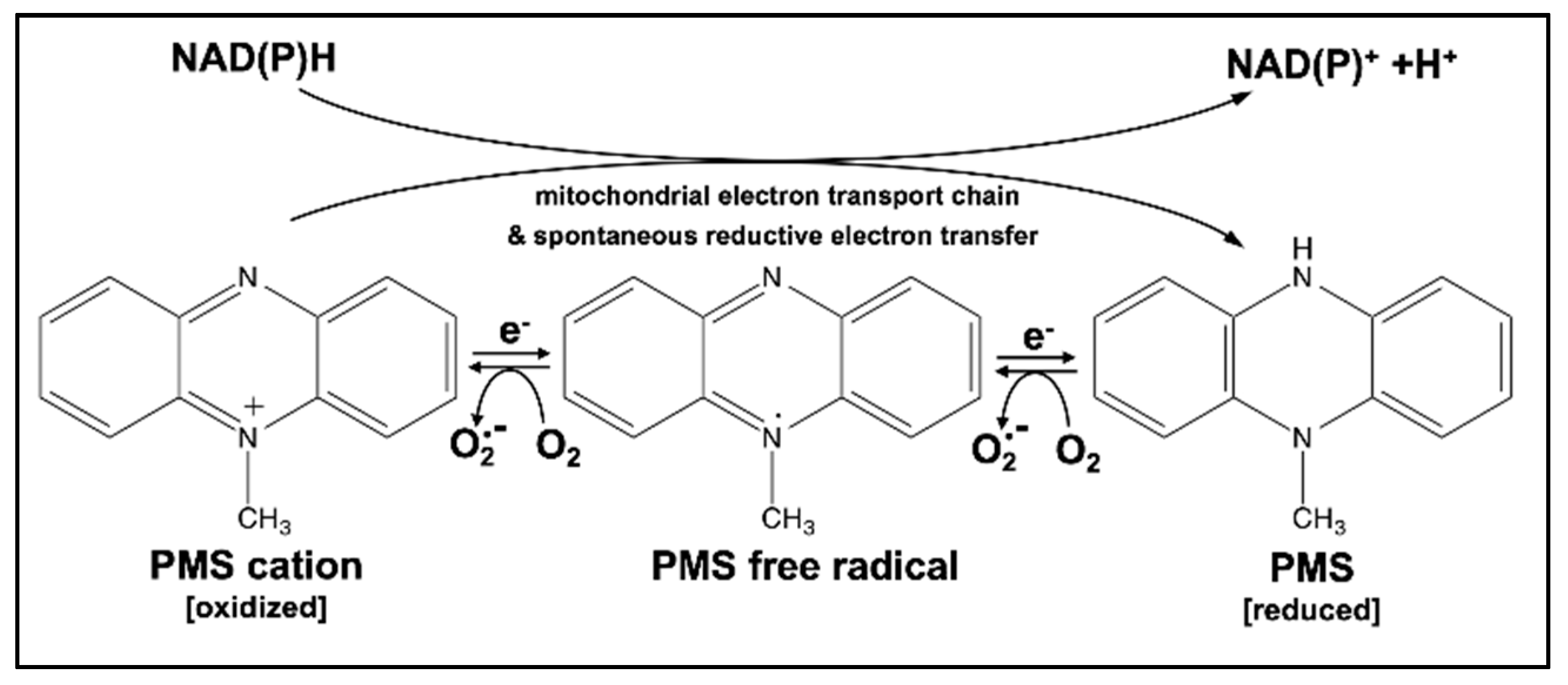
1. Introduction
2. Results
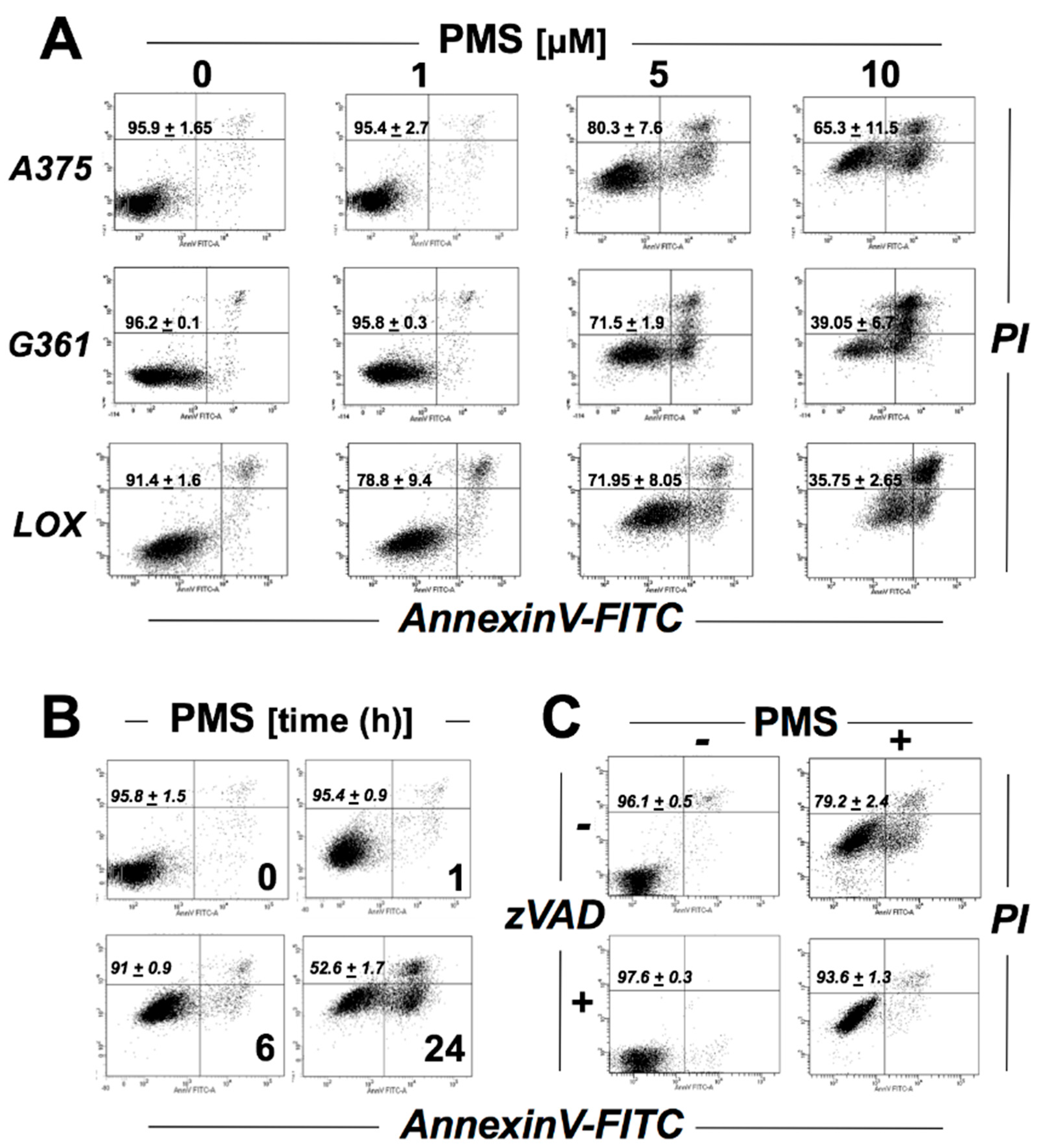
2.1. PMS Induces Caspase-Dependent Death in A375 Malignant Melanoma Cells
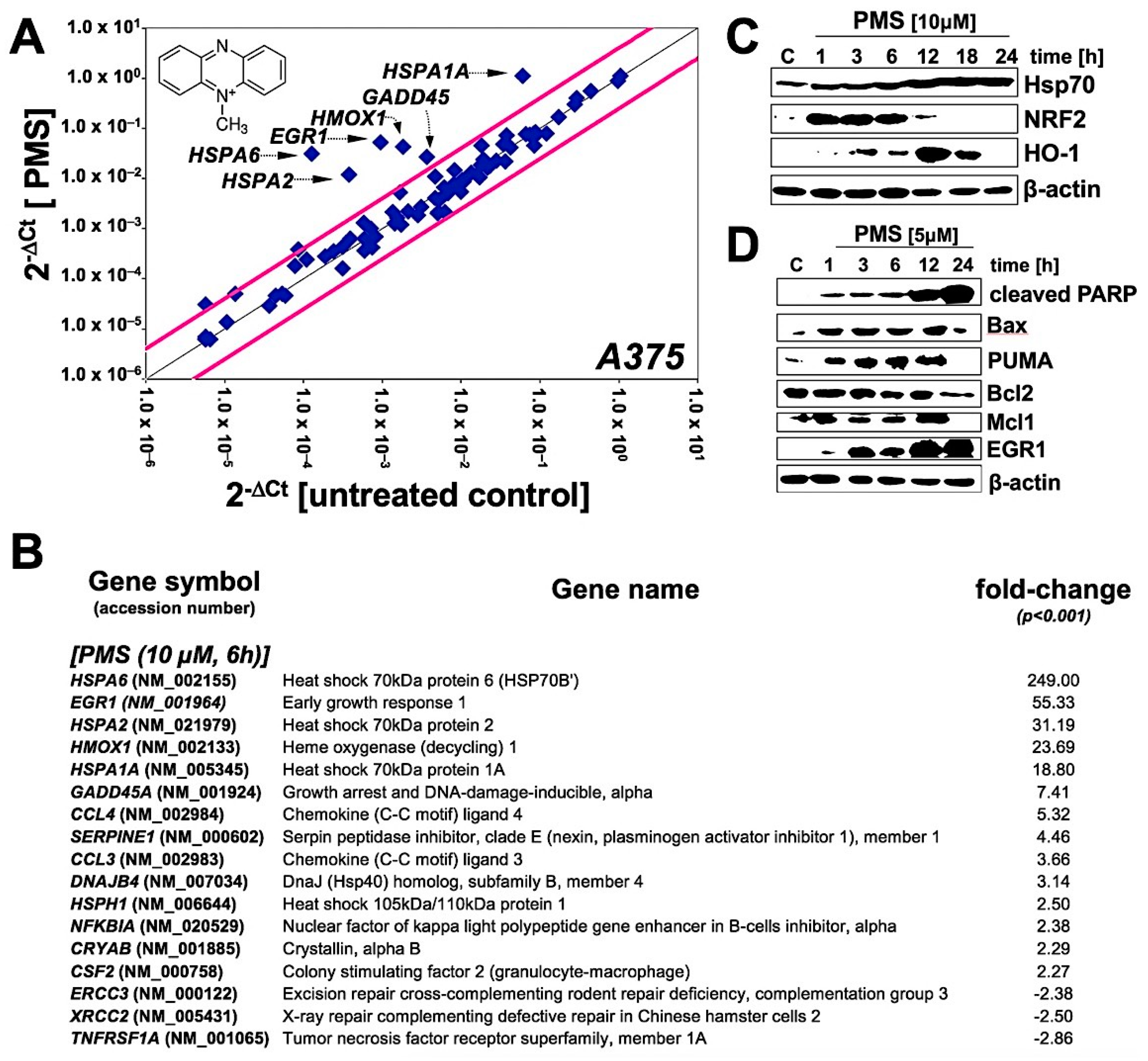
2.2. Array Analysis Reveals Upregulation of Heat Shock, Oxidative, and Genotoxic Stress Response Gene Expression in PMS-Exposed A375 Malignant Melanoma Cells
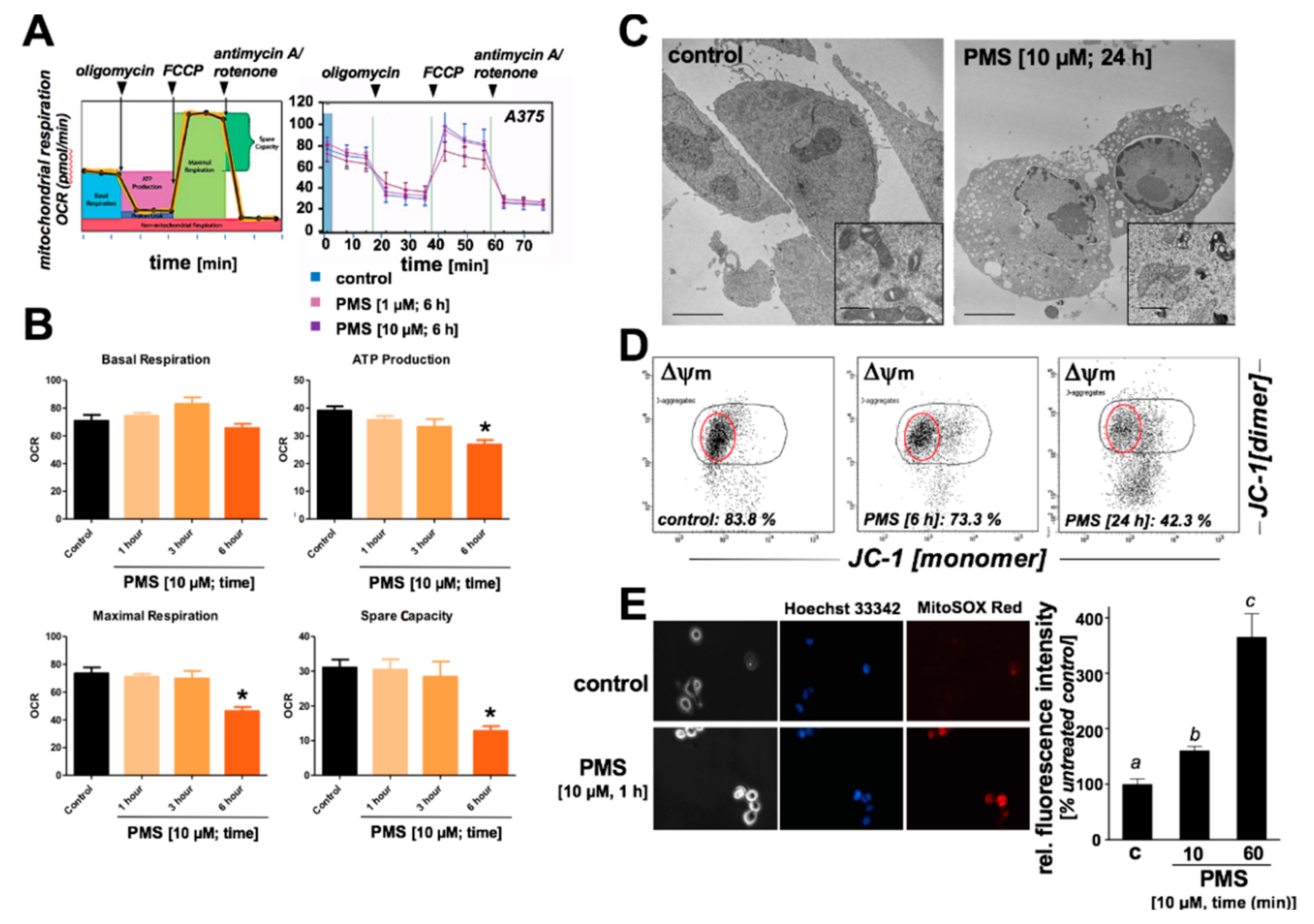
2.3. PMS Induces Mitochondriotoxicity in Human A375 Malignant Melanoma Cells Characterized by Ultrastructural Changes, Mitochondrial Respiratory Impairment, Loss of Transmembrane Potential, and Superoxide Production
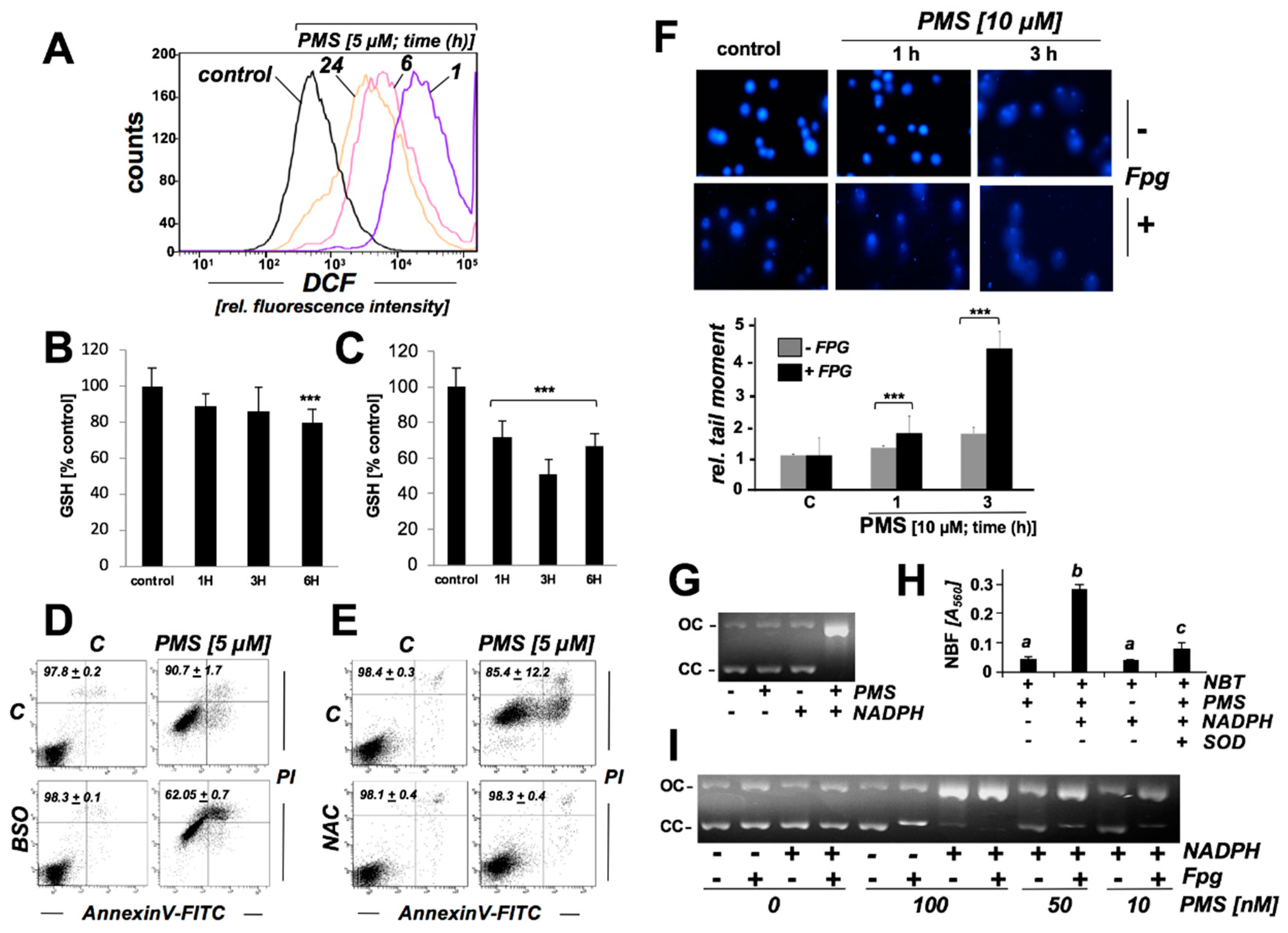
2.4. PMS Induces Oxidative Stress in Human A375 Malignant Melanoma Cells Associated with Glutathione Depletion And Impairment Of Genomic Integrity
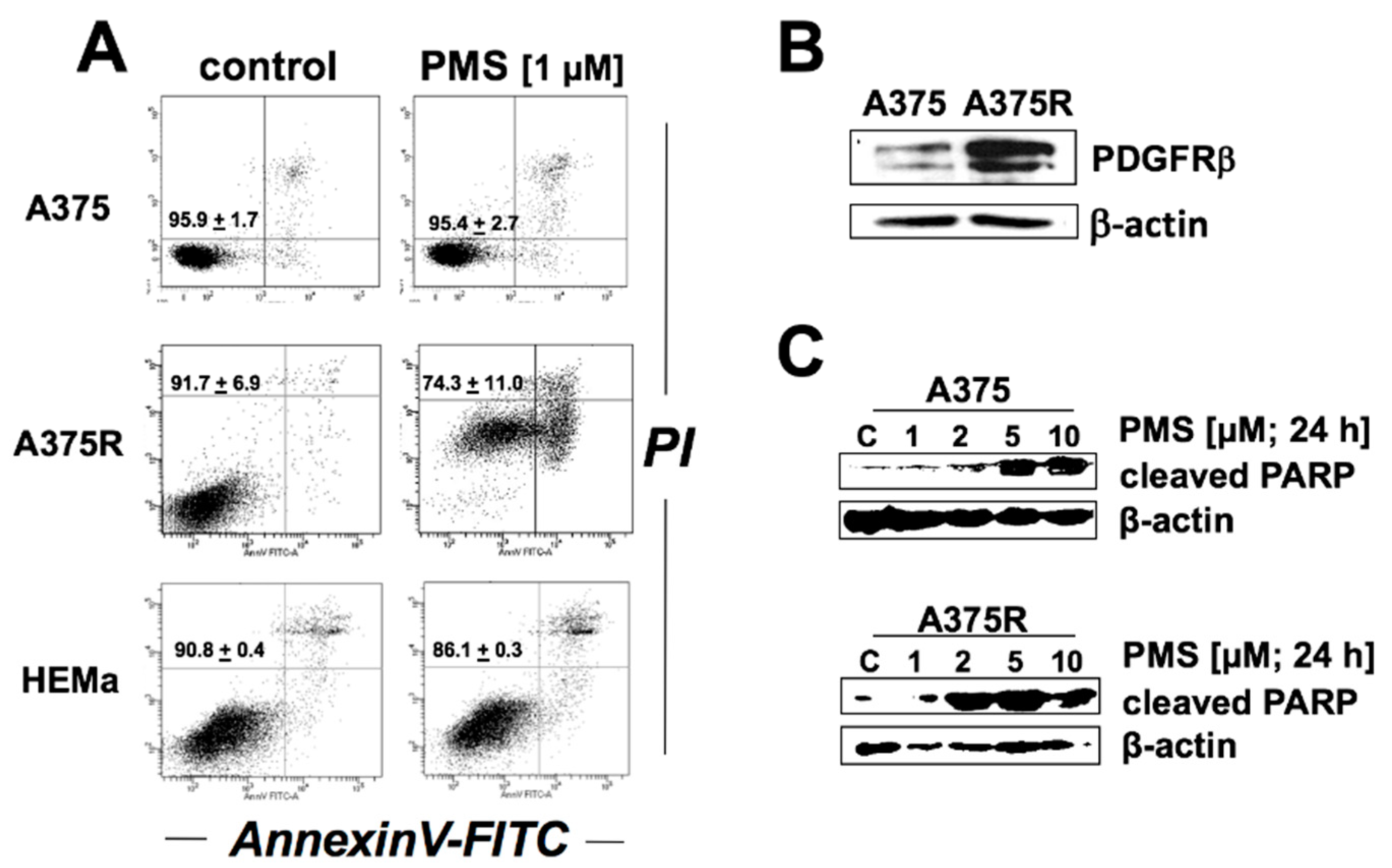
2.5. PMS Preferentially Induces Cell Death in Vemurafenib-Resistant A375 Melanoma Cells (A375R)
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Flow Cytometry Analysis of Cell Viability
4.4. Human Stress and Toxicity Pathfinder RT2ProfilerTM PCR Expression Array
4.5. Immunoblot Detection
4.6. PMS-Modulation of Mitochondrial Respiration in a375 Melanoma Cells
4.7. Detection of Intracellular Oxidative Stress by Flow Cytometric Analysis
4.8. MitoSOX Fluorescence Imaging
4.9. Mitochondrial Transmembrane Potential
4.10. Determination of Reduced Cellular Glutathione Content
4.11. Transmission Electron Microscopy
4.12. Comet Assay (Alkaline Single-Cell Gel Electrophoresis)
4.13. Plasmid Cleavage Assay
4.14. NBT Assay for PMS-Dependent Superoxide Formation
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., 3rd; Wondrak, G.T. Experimental therapeutics: Targeting the redox Achilles heel of cancer. Curr. Opin. Investig. Drugs 2007, 8, 1022–1037. [Google Scholar] [PubMed]
- Fantin, V.R.; Berardi, M.J.; Scorrano, L.; Korsmeyer, S.J.; Leder, P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell 2002, 2, 29–42. [Google Scholar] [CrossRef]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; Andre, F.; Jonneaux, A.; Scalbert, C.; Garcon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.A. Is reliance on mitochondrial respiration a “chink in the armor” of therapy-resistant cancer? Cancer Cell 2014, 26, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B., 3rd; Qiao, S.; Azimian, S.; Lesson, J.L.; Wondrak, G.T. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest. New Drugs 2012, 30, 1289–1301. [Google Scholar] [CrossRef]
- Zaugg, W.S. Spectroscopic Characteristics and Some Chemical Properties of N-Methylphenazinium Methyl Sulfate (Phenazine Methosulfate) and Pyocyanine at the Semiquidnoid Oxidation Level. J. Biol. Chem. 1964, 239, 3964–3970. [Google Scholar]
- Hassan, H.M.; Fridovich, I. Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch. Biochem. Biophys. 1979, 196, 385–395. [Google Scholar] [CrossRef]
- Gardner, P.R.; Raineri, I.; Epstein, L.B.; White, C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 1995, 270, 13399–13405. [Google Scholar] [CrossRef]
- Carloni, P.; Greci, L.; Maurelli, E.; Stipa, P.; Wozniak, M.; Marrosu, G.; Petrucci, R.; Rtrazza, A. The role of oxygen in the reduction of tetrazolium salts with NADH mediated by 5-methyl phenazonium methylsulfate. An EPR and voltammetric study. Res. Chem. Intermed. 1993, 19, 643–656. [Google Scholar] [CrossRef]
- Sinha, S.; Shen, X.; Gallazzi, F.; Li, Q.; Zmijewski, J.W.; Lancaster, J.R., Jr.; Gates, K.S. Generation of Reactive Oxygen Species Mediated by 1Hydroxyphenazine, a Virulence Factor of Pseudomonas aeruginosa. Chem. Res. Toxicol. 2015, 28, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Manago, A.; Becker, K.A.; Carpinteiro, A.; Wilker, B.; Soddemann, M.; Seitz, A.P.; Edwards, M.J.; Grassme, H.; Szabo, I.; Gulbins, E. Pseudomonas aeruginosa pyocyanin induces neutrophil death via mitochondrial reactive oxygen species and mitochondrial acid sphingomyelinase. Antioxid. Redox Signal. 2015, 22, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Justiniano, R.; Williams, J.D.; Cabello, C.M.; Qiao, S.; Wondrak, G.T. The Tryptophan-Derived Endogenous Aryl Hydrocarbon Receptor Ligand 6-Formylindolo [3,2-b]Carbazole Is a Nanomolar UVA Photosensitizer in Epidermal Keratinocytes. J. Invest. Dermatol. 2015, 135, 1649–1658. [Google Scholar] [CrossRef]
- Justiniano, R.; Williams, J.D.; Perer, J.; Hua, A.; Lesson, J.; Park, S.L.; Wondrak, G.T. The B6 -vitamer Pyridoxal is a Sensitizer of UVA-induced Genotoxic Stress in Human Primary Keratinocytes and Reconstructed Epidermis. Photochem. Photobiol. 2017, 93, 990–998. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Jacobson, E.L.; Jacobson, M.K. Photosensitization of DNA damage by glycated proteins. Photochem. Photobiol. Sci. 2002, 1, 355–363. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef]
- Livingstone, E.; Swann, S.; Lilla, C.; Schadendorf, D.; Roesch, A. Combining BRAF(V) (600E) inhibition with modulators of the mitochondrial bioenergy metabolism to overcome drug resistance in metastatic melanoma. Exp. Dermatol. 2015, 24, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic-Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wicha, M.S. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin. Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Cabello, C.M.; Bair, W.B., 3rd; Bause, A.S.; Wondrak, G.T. Antimelanoma activity of the redox dye DCPIP (2,6-dichlorophenolindophenol) is antagonized by NQO1. Biochem. Pharmacol. 2009, 78, 344–354. [Google Scholar] [CrossRef]
- Hou, G.X.; Liu, P.P.; Zhang, S.; Yang, M.; Liao, J.; Yang, J.; Hu, Y.; Jiang, W.Q.; Wen, S.; Huang, P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Santhosh, S.M.; Coppo, L.; Ogata, F.T.; Lu, J.; Holmgren, A. The combination of ascorbate and menadione causes cancer cell death by oxidative stress and replicative stress. Free Radic. Biol. Med. 2019, 134, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lu, M.C.; You, Q.D. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Inhibition: An Emerging Strategy in Cancer Therapy. J. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Magda, D.; Miller, R.A. Motexafin gadolinium: A novel redox active drug for cancer therapy. Semin. Cancer Biol. 2006, 16, 466–476. [Google Scholar] [CrossRef]
- Wondrak, G.T. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apoptosis. Free Radic. Biol. Med. 2007, 43, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Eigentler, T.K.; Keilholz, U.; Hauschild, A.; Kirkwood, J.M. Systematic review of medical treatment in melanoma: Current status and future prospects. Oncologist 2011, 16, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Luke, J.J.; Hodi, F.S. Ipilimumab, vemurafenib, dabrafenib, and trametinib: Synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist 2013, 18, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Davies, M.A. Targeted therapy for melanoma: Rational combinatorial approaches. Oncogene 2014, 33, 1–9. [Google Scholar] [CrossRef]
- Fried, L.; Arbiser, J.L. The reactive oxygen-driven tumor: Relevance to melanoma. Pigment Cell Melanoma Res. 2008, 21, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T. Melanomagenic Gene Alterations Viewed from a Redox Perspective: Molecular Mechanisms and Therapeutic Opportunities. In Stress Response Pathways in Cancer; Springer: Heidelberg, Germany; New York, NY, USA, 2015; Volume 1, pp. 285–309. ISBN 978-94-017-9420-6. [Google Scholar]
- Acquaviva, J.; Smith, D.L.; Jimenez, J.P.; Zhang, C.; Sequeira, M.; He, S.; Sang, J.; Bates, R.C.; Proia, D.A. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol. Cancer Ther. 2014, 13, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Egger, M.E.; McMasters, K.M.; Hao, H. Differential expression of ABCB5 in BRAF inhibitor-resistant melanoma cell lines. BMC Cancer 2018, 18, 675. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.L.; Qiao, S.; Lesson, J.L.; Rojo de la Vega, M.; Park, S.L.; Seanez, C.M.; Gokhale, V.; Cabello, C.M.; Wondrak, G.T. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J. Biol. Chem. 2015, 290, 1623–1638. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front Neurosci. 2017, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Justiniano, R.; Perer, J.; Hua, A.; Fazel, M.; Krajisnik, A.; Cabello, C.M.; Wondrak, G.T. A Topical Zinc Ionophore Blocks Tumorigenic Progression in UV-exposed SKH-1 High-risk Mouse Skin. Photochem. Photobiol. 2017, 93, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Tao, S.; Rojo de la Vega, M.; Park, S.L.; Vonderfecht, A.A.; Jacobs, S.L.; Zhang, D.D.; Wondrak, G.T. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy 2013, 9, 2087–2102. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, A.B.; Justiniano, R.; Perer, J.; Park, S.L.; Li, H.; Cabello, C.M.; Wondrak, G.T. Repurposing the Electron Transfer Reactant Phenazine Methosulfate (PMS) for the Apoptotic Elimination of Malignant Melanoma Cells through Induction of Lethal Oxidative and Mitochondriotoxic Stress. Cancers 2019, 11, 590. https://doi.org/10.3390/cancers11050590
Hua AB, Justiniano R, Perer J, Park SL, Li H, Cabello CM, Wondrak GT. Repurposing the Electron Transfer Reactant Phenazine Methosulfate (PMS) for the Apoptotic Elimination of Malignant Melanoma Cells through Induction of Lethal Oxidative and Mitochondriotoxic Stress. Cancers. 2019; 11(5):590. https://doi.org/10.3390/cancers11050590
Chicago/Turabian StyleHua, Anh B., Rebecca Justiniano, Jessica Perer, Sophia L. Park, Hui Li, Christopher M. Cabello, and Georg T. Wondrak. 2019. "Repurposing the Electron Transfer Reactant Phenazine Methosulfate (PMS) for the Apoptotic Elimination of Malignant Melanoma Cells through Induction of Lethal Oxidative and Mitochondriotoxic Stress" Cancers 11, no. 5: 590. https://doi.org/10.3390/cancers11050590
APA StyleHua, A. B., Justiniano, R., Perer, J., Park, S. L., Li, H., Cabello, C. M., & Wondrak, G. T. (2019). Repurposing the Electron Transfer Reactant Phenazine Methosulfate (PMS) for the Apoptotic Elimination of Malignant Melanoma Cells through Induction of Lethal Oxidative and Mitochondriotoxic Stress. Cancers, 11(5), 590. https://doi.org/10.3390/cancers11050590