CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Anti-Tumor Reagents
2.3. Transfection and Establishment of EGFR−/− Cell Lines
2.4. Western Blot
2.5. DNA Isolation, Polymerase Chain Reaction (PCR), and Sequencing
2.6. Colony Formation Assay
2.7. Wound Healing Assay
2.8. Cell Migration Assay
2.9. MTS Assay
2.10. Flow Cytometric Analysis
2.11. RNA Isolation and Quantitative Reverse Transcriptase PCR (qRT-PCR)
2.12. CXCR7 Knockdown Using siRNAs
2.13. EGFR Rescue in A549 EGFR−/− Cells
2.14. Patient Tumor Samples and Immunohistochemistry (IHC)
2.15. Statistical Analysis
3. Results
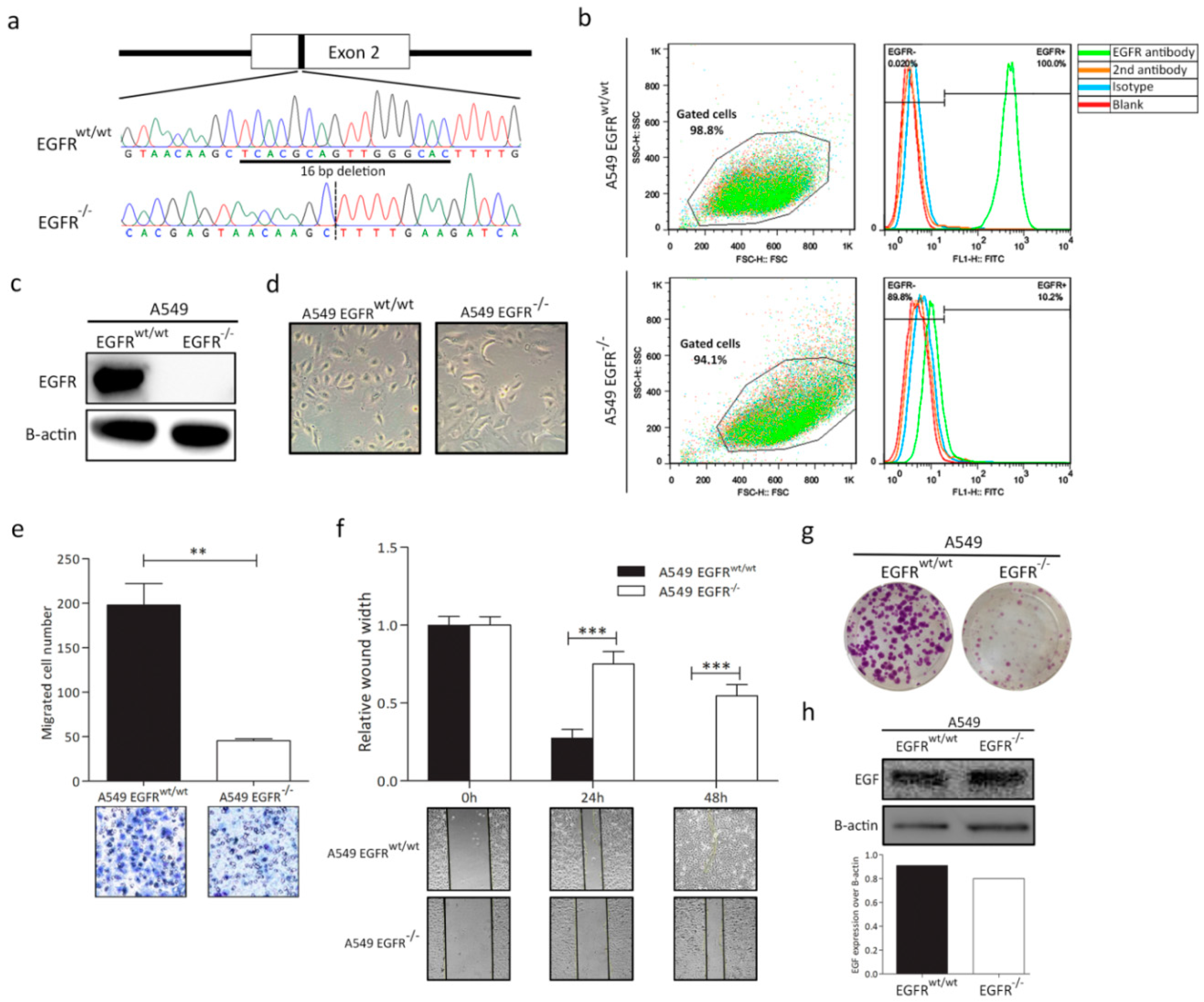
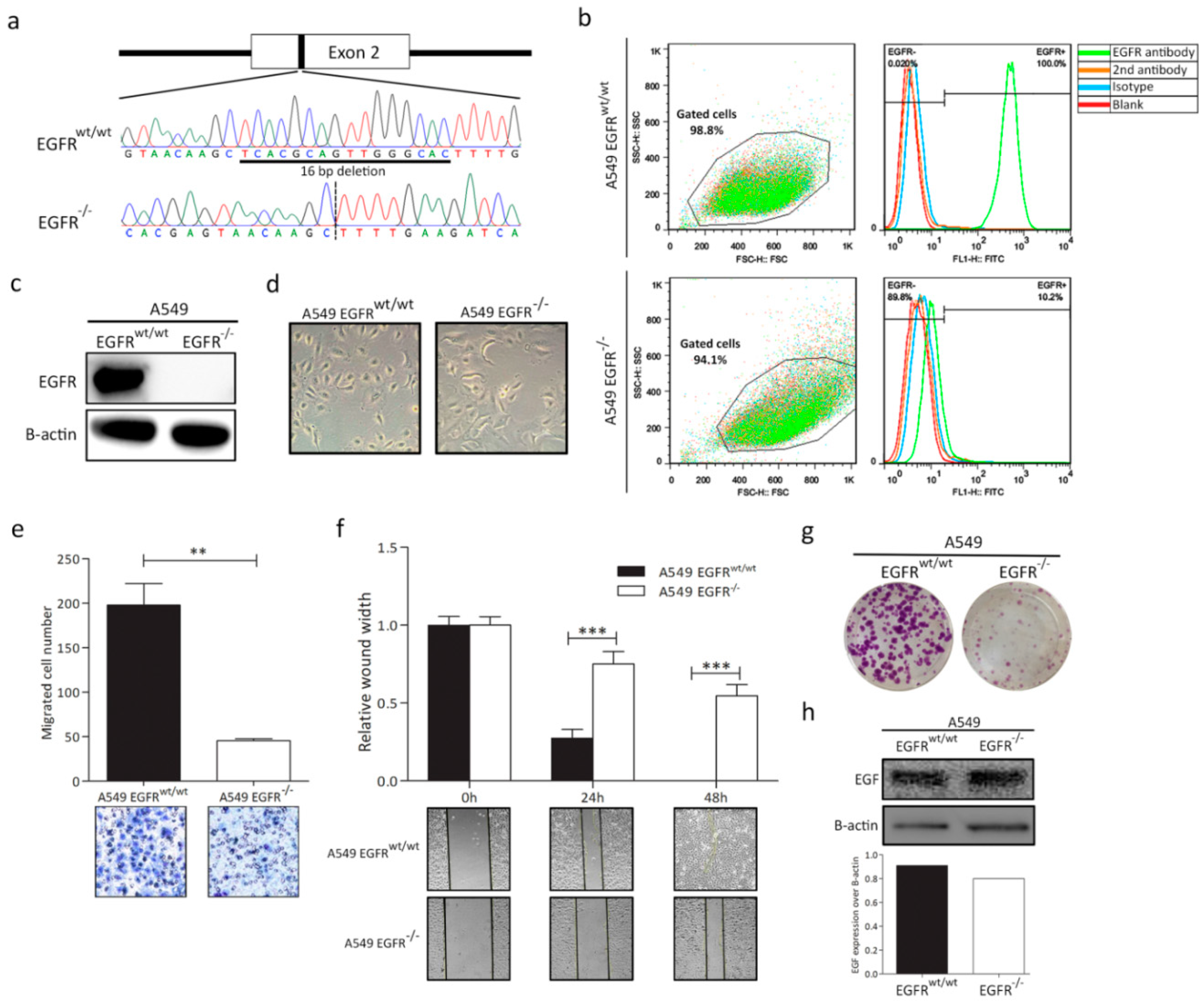
3.1. EGFR Knockout in A549 Cell Line
3.2. EGFR Plays a Significant Role in Cell Progression in KRAS-Dependent Lung Cancer Cells
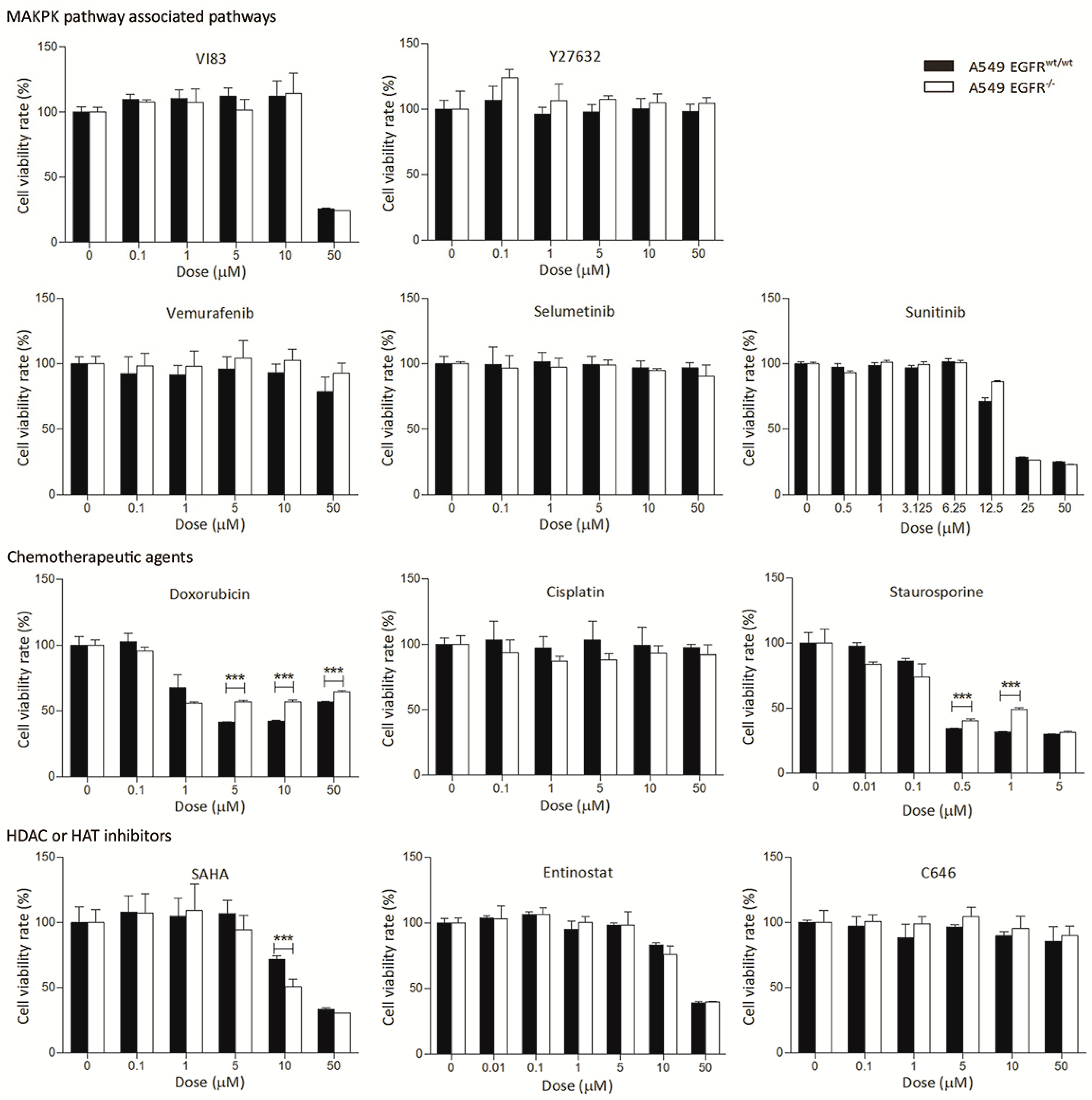
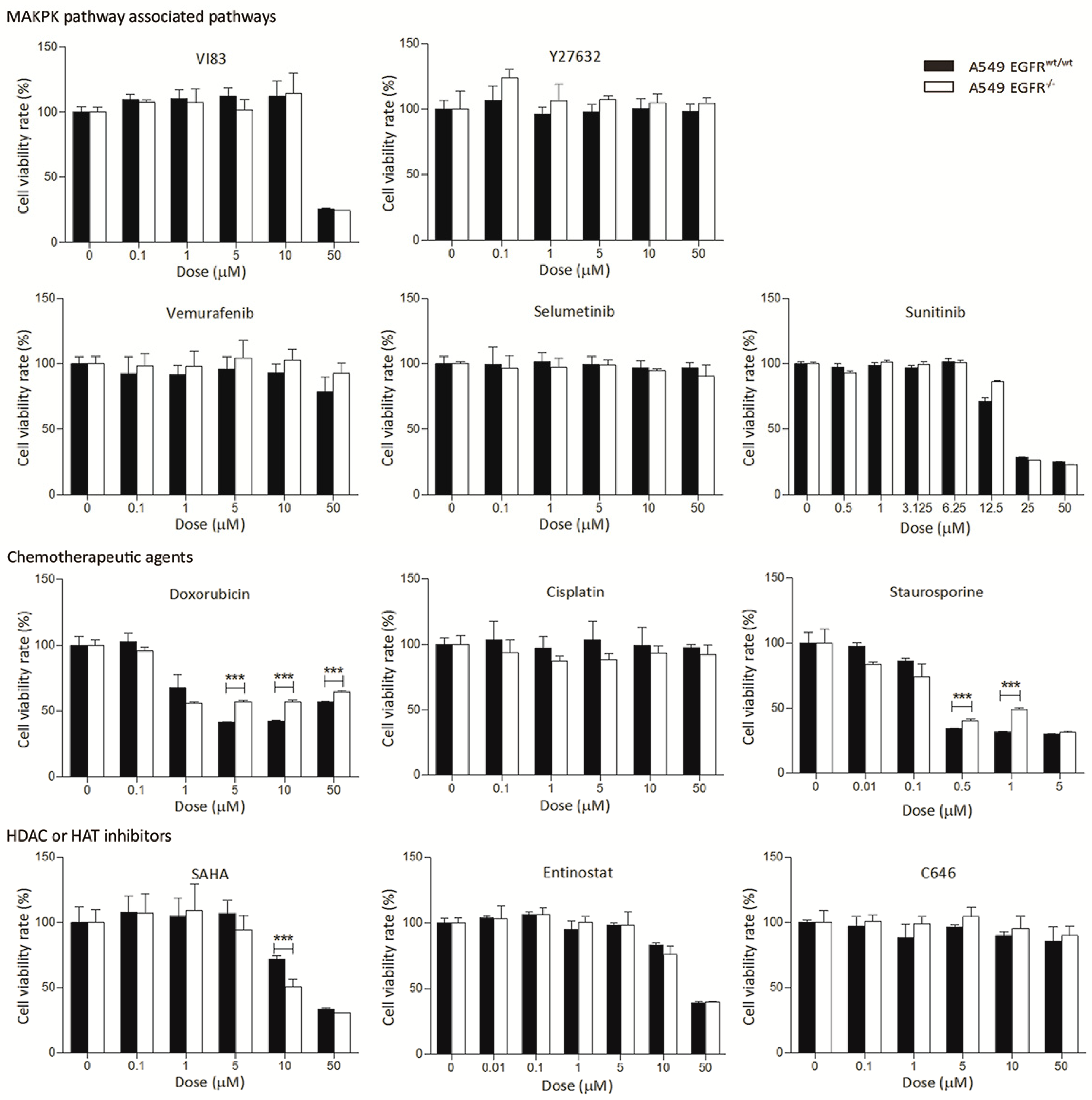
3.3. EGFR Knockout Does Not Significantly Affect Drug Sensitivity
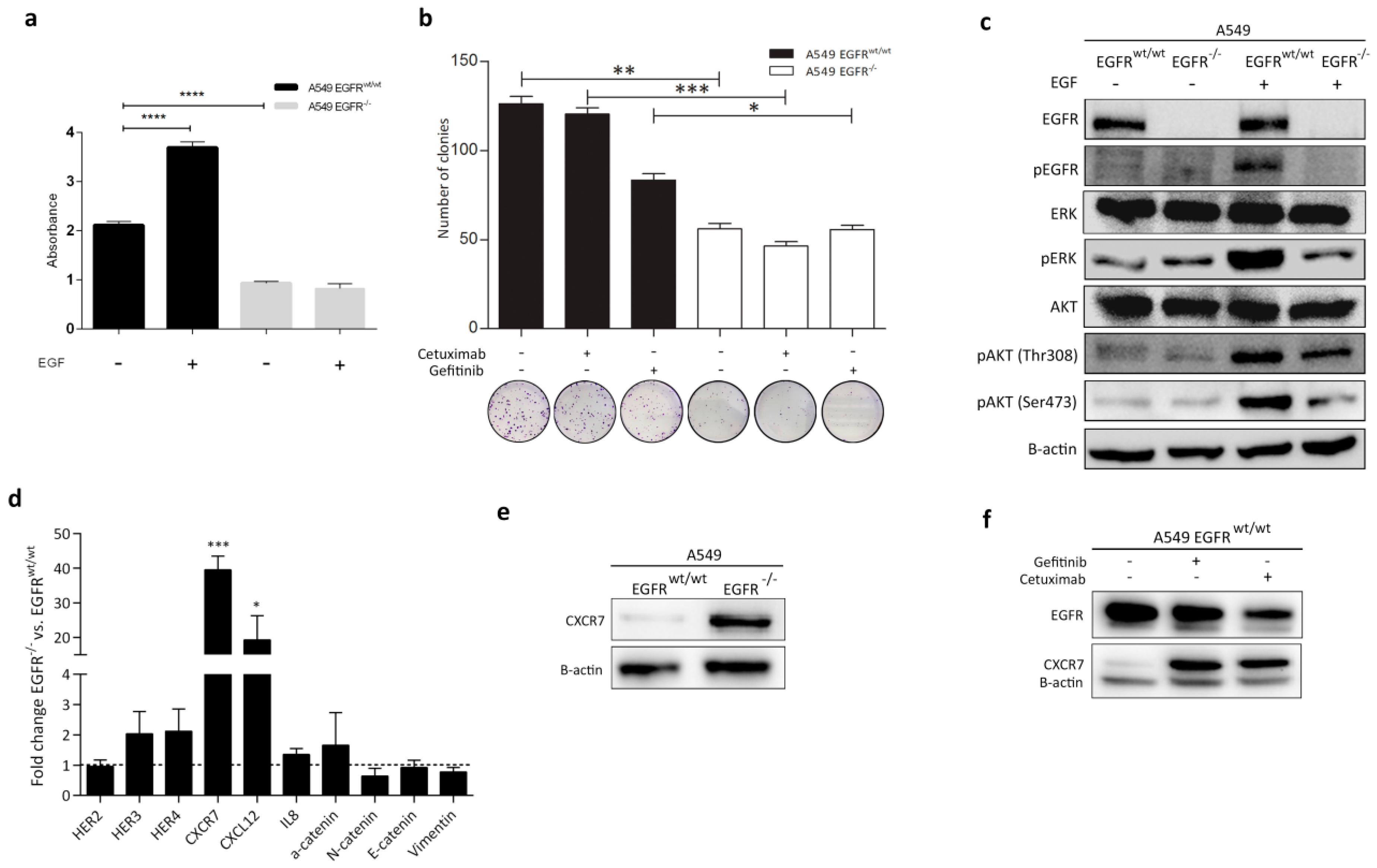
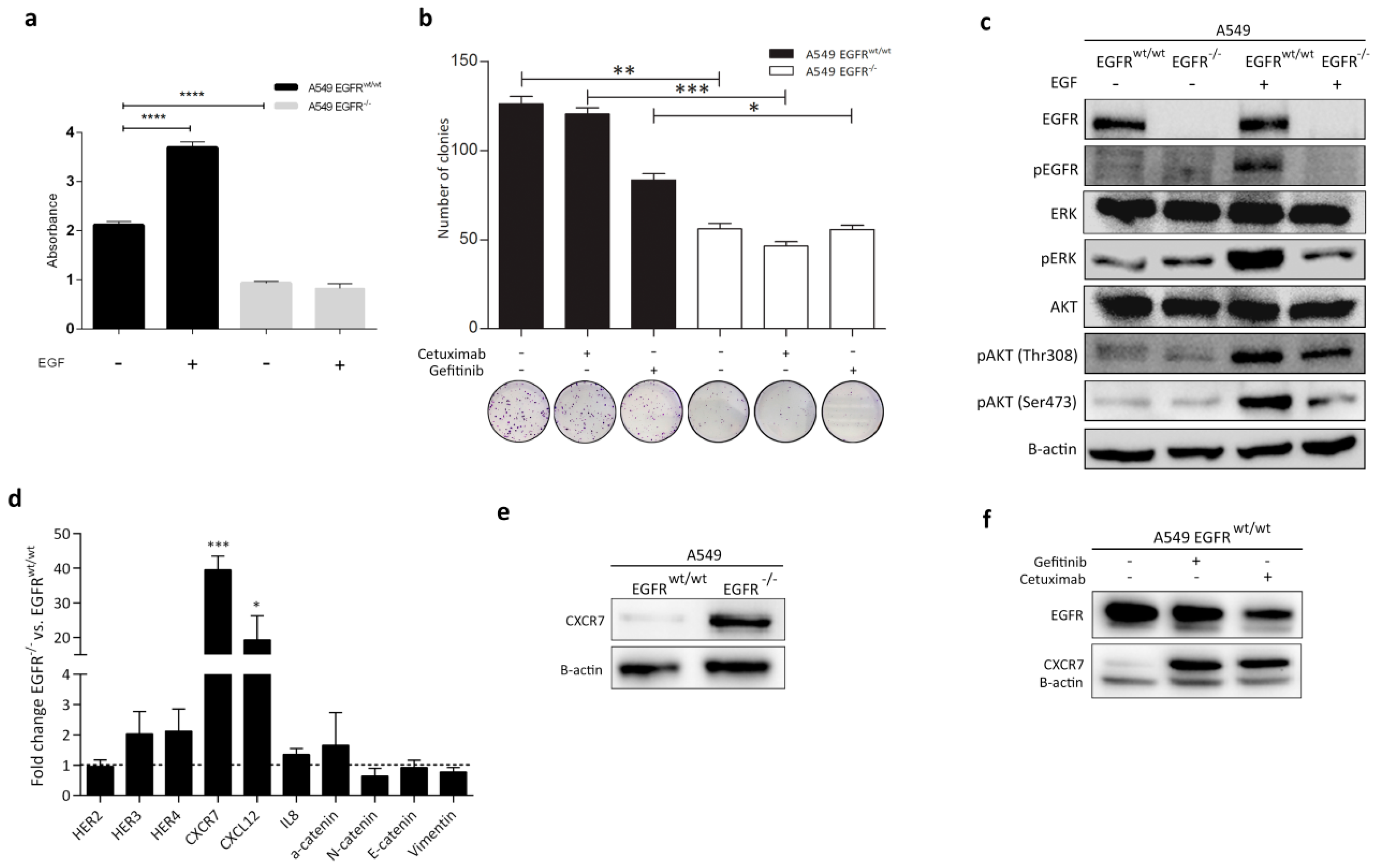
3.4. Effect of EGFR Loss on Downstream Pathways
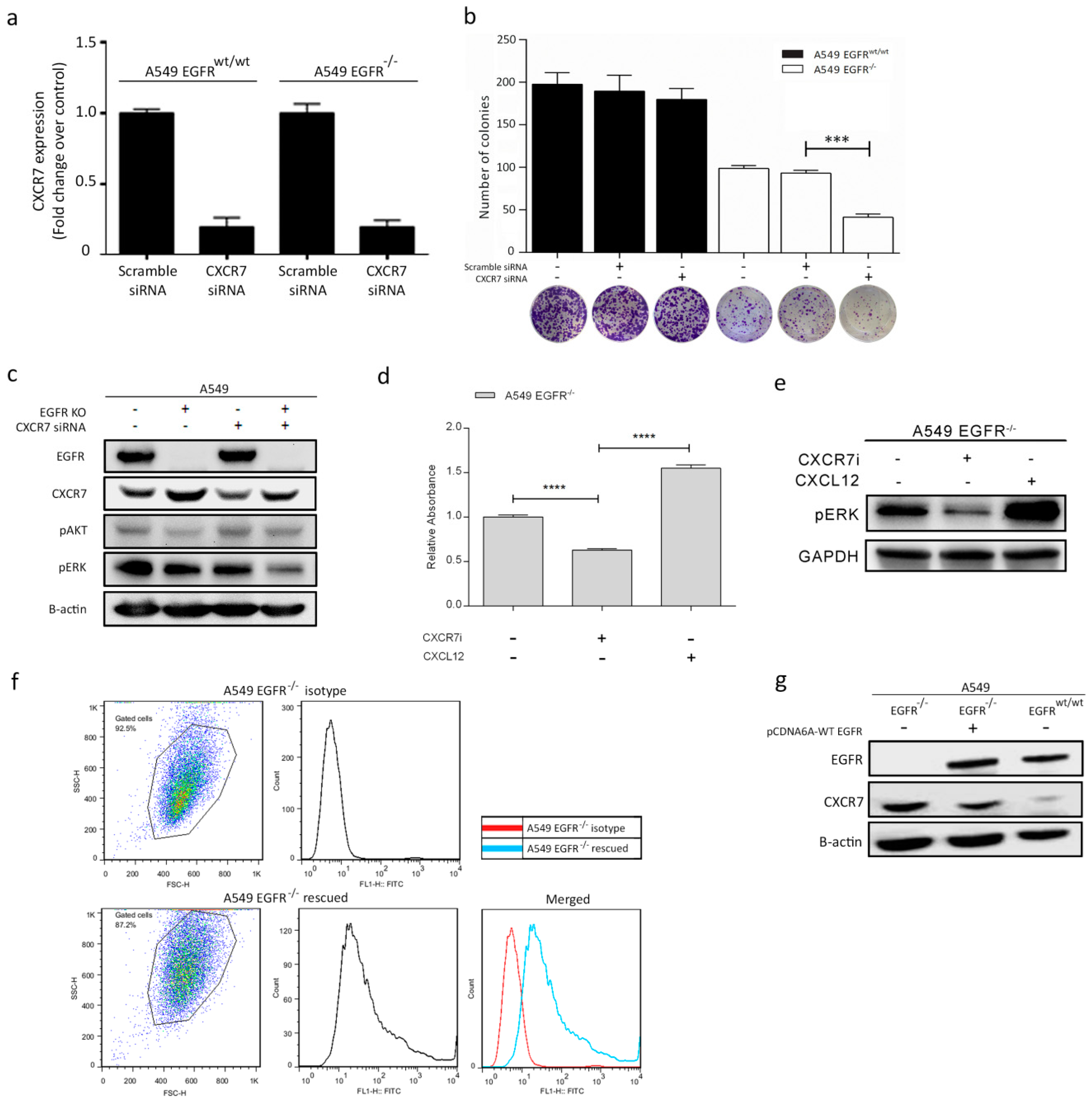
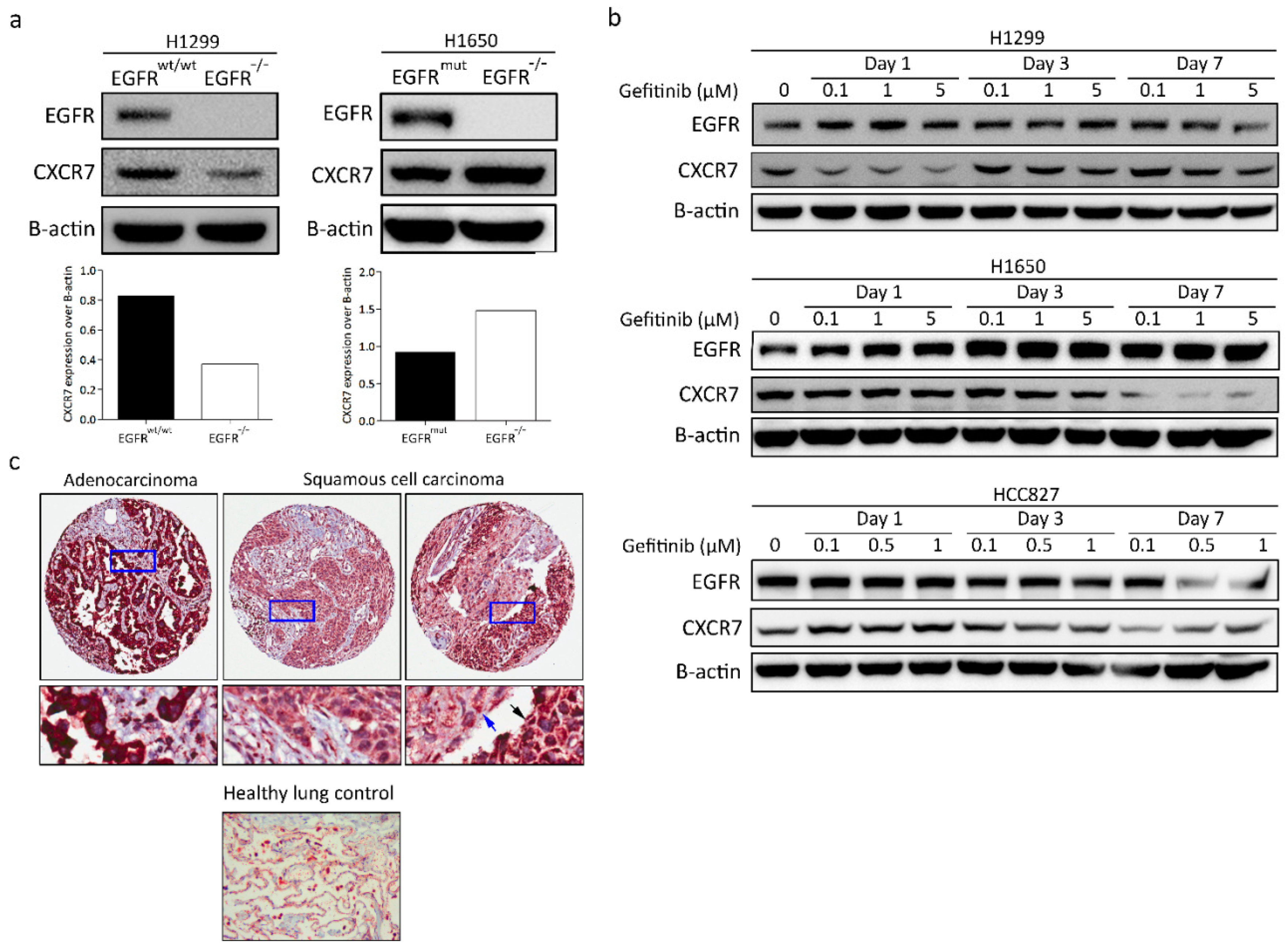
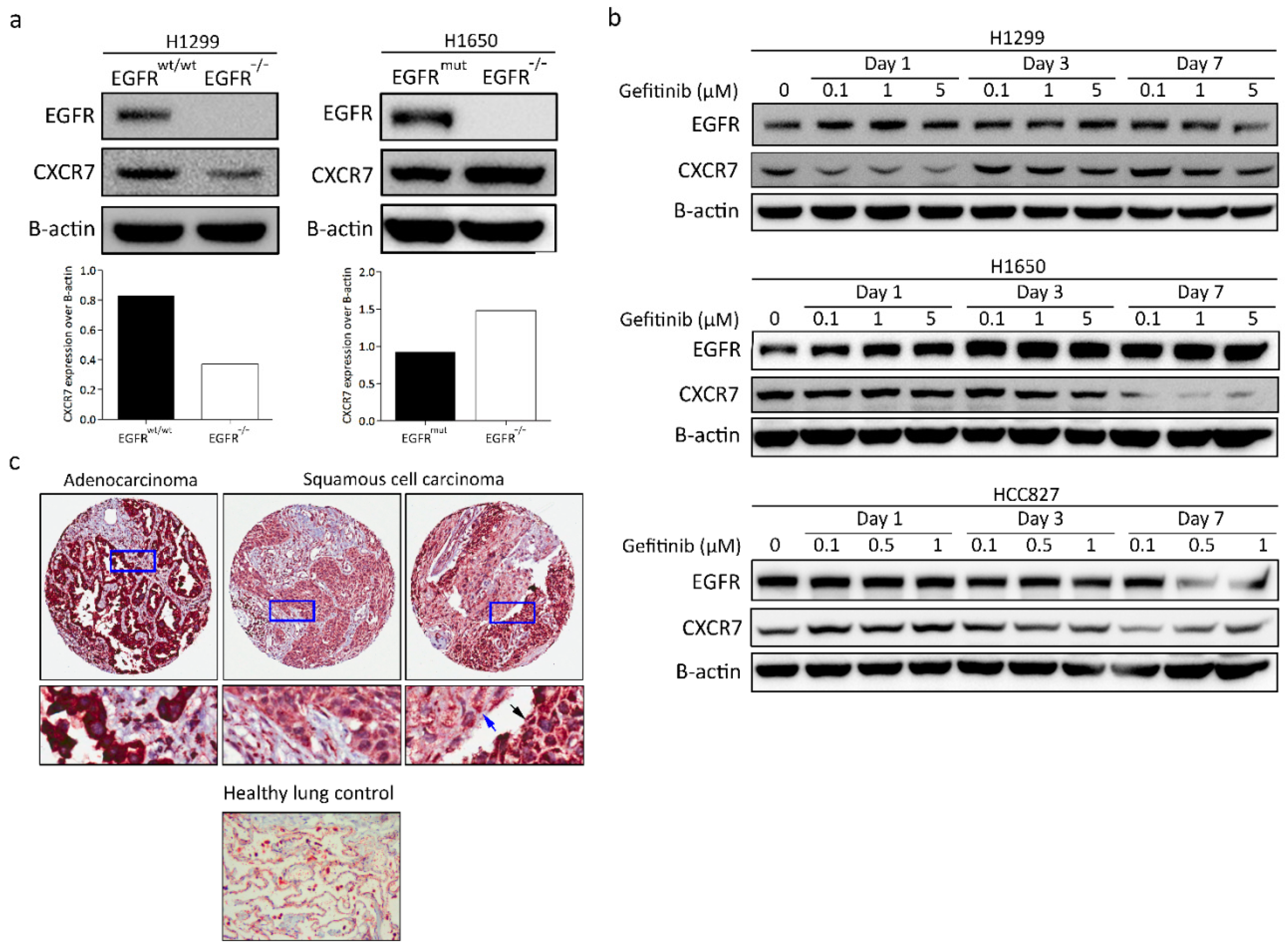
3.5. CXCR7 Is Significantly Upregulated in A549 EGFR−/− Cells and EGFRwt/wt Cells Treated with EGFR Inhibitors
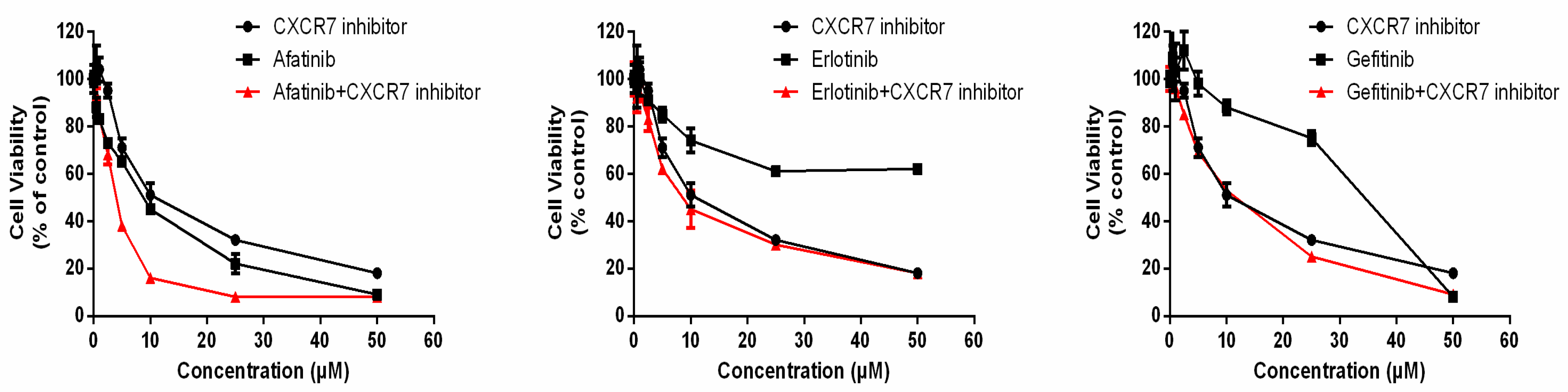
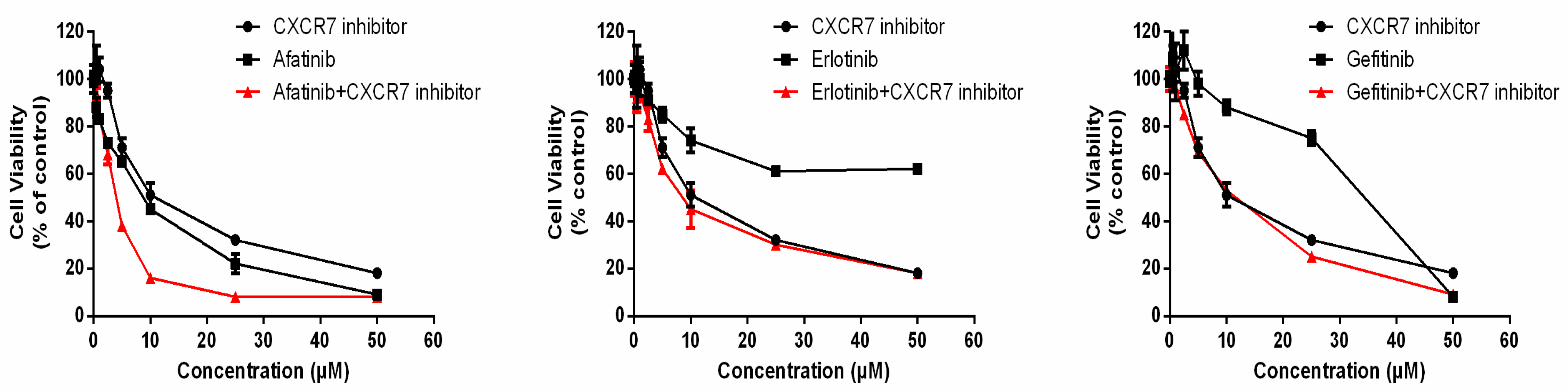
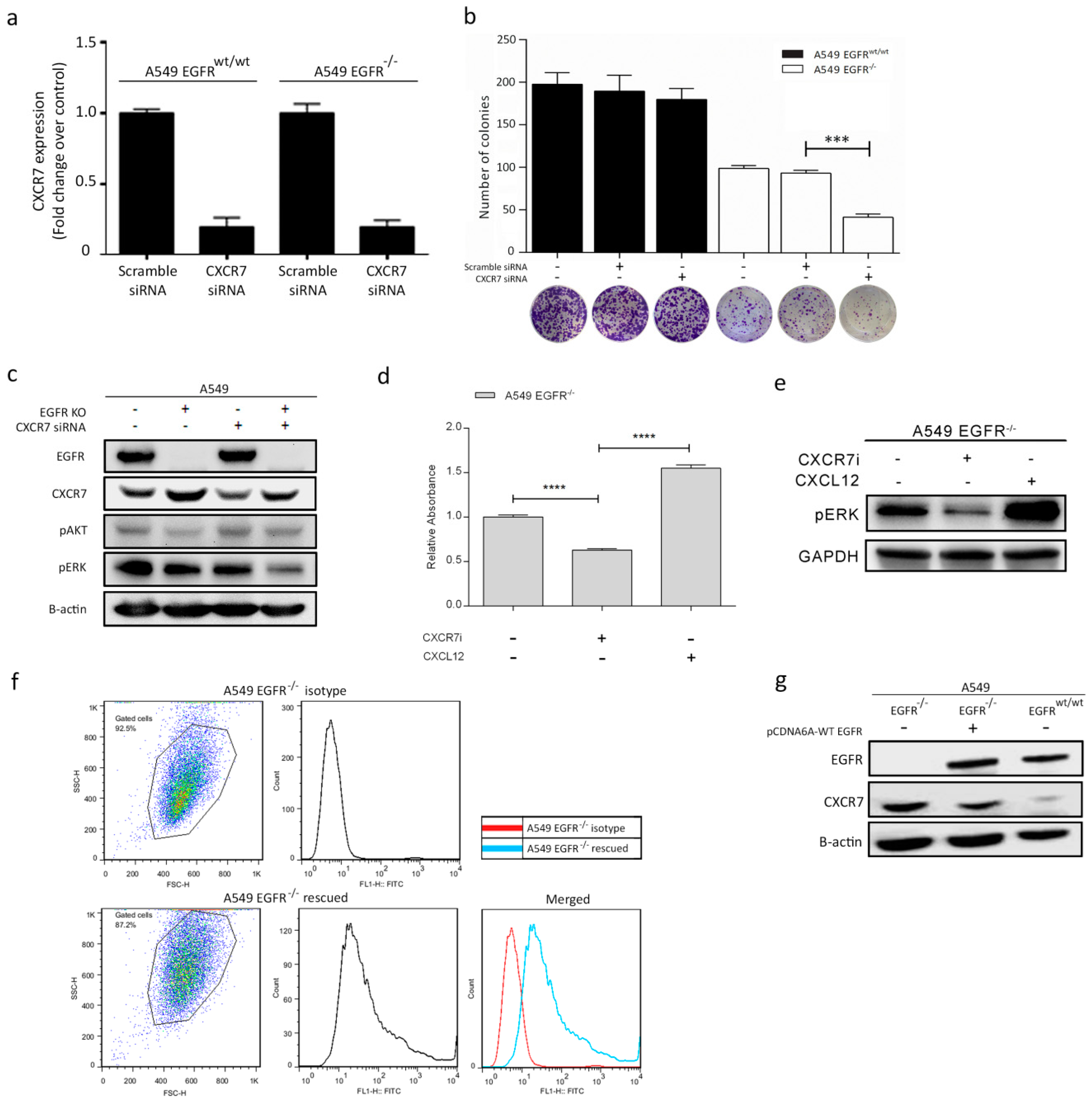
3.6. Dual Inhibition of CXCR7 and EGFR Downregulates MAPK (ERK1/2) and Suppresses Proliferation
3.7. Rescue of EGFR Leads to CXCR7 Downregulation in A549 EGFR−/− Cells
3.8. CXCR7 Expression, Altered by the Ablation of EGFR, Is Associated with the Genotype of NSCLC
3.9. CXCR7 Is Highly Expressed in Primary Lung Adenocarcinoma
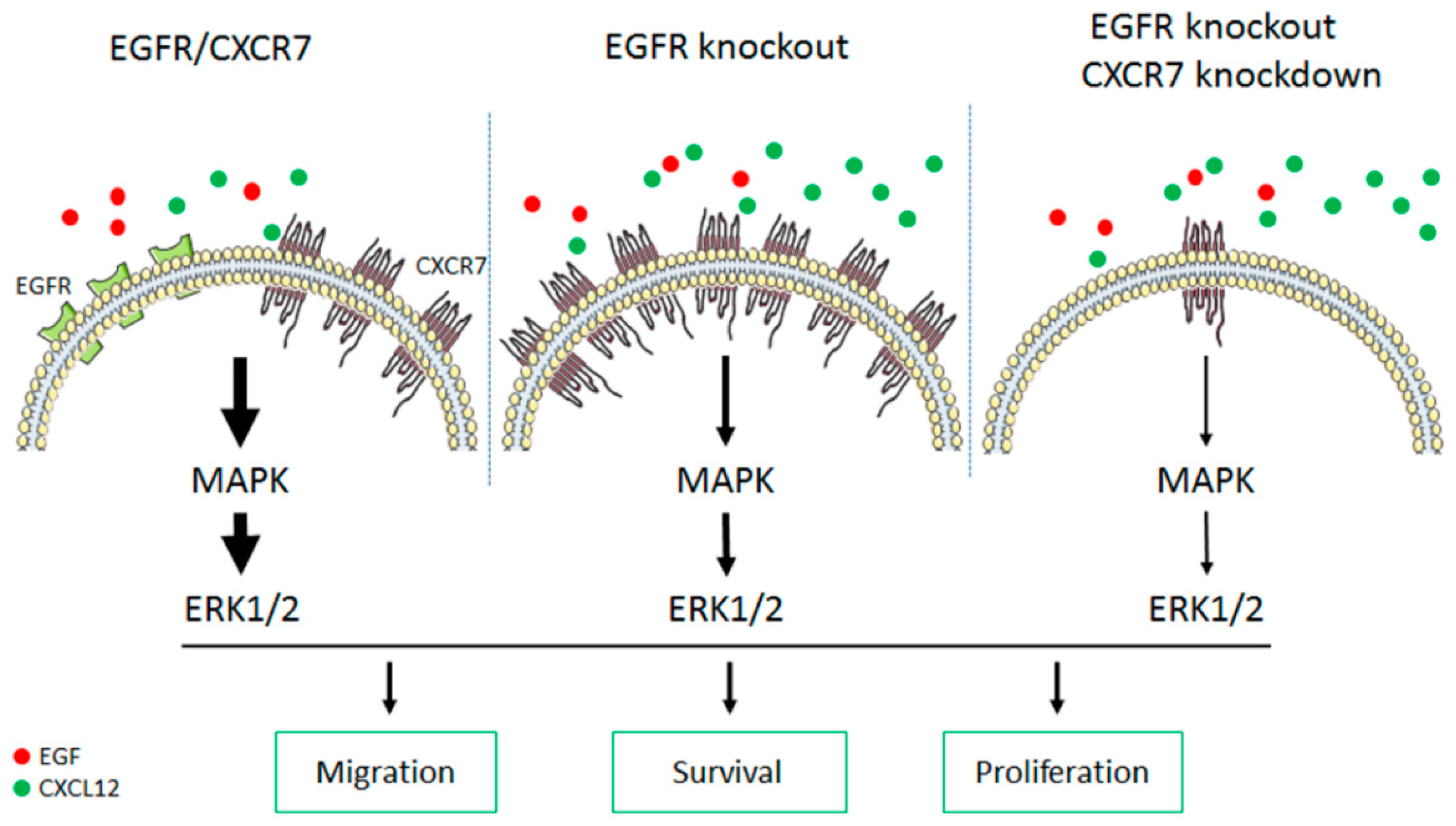
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [Green Version]
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR Mutations in Non–Small-Cell Lung Cancer: Analysis of a Large Series of Cases and Development of a Rapid and Sensitive Method for Diagnostic Screening with Potential Implications on Pharmacologic Treatment. J. Clin. Oncol. 2005, 23, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Hahn, S.; Kim, D.W.; Suh, K.J.; Keam, B.; Kim, T.M.; Lee, S.H.; Heo, D.S. Epidermal growth factor receptor tyrosine kinase inhibitors vs conventional chemotherapy in non–small cell lung cancer harboring wild-type epidermal growth factor receptor: A meta-analysis. JAMA 2014, 311, 1430–1437. [Google Scholar] [CrossRef]
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife 2017, 6, e18970. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK Signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saber, A.; Liu, B.; Ebrahimi, P.; Haisma, H.J. CRISPR/Cas9 for overcoming drug resistance in solid tumors. DARU J. Pharm. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Saber, A.; Haisma, H.J. CRISPR/Cas9: A powerful tool for identification of new targets for cancer treatment. Drug Discov. Today 2019. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, H.; Moore, J.; Malonia, S.K.; Li, Y.; Ozata, D.M.; Hough, S.; Song, C.-Q.; Smith, J.L.; Fischer, A.; Weng, Z.; et al. Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 3648–3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. 2008, 13, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, L.; Sánchez-Mateos, P.; Cabañas, C. CXCR7 impact on CXCL12 biology and disease. Trends Mol. Med. 2013, 19, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libert, F.; Parmentier, M.; Lefort, A.; Dinsart, C.; Van Sande, J.; Maenhaut, C.; Simons, M.J.; Dumont, J.E.; Vassart, G. Selective amplification and cloning of four new members of the G protein-coupled receptor family. Science 1989, 244, 569–572. [Google Scholar] [CrossRef]
- Sreedharan, S.P.; Robichon, A.; Peterson, K.E.; Goetzl, E.J. Cloning and expression of the human vasoactive intestinal peptide receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 4986–4990. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Muller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [Green Version]
- Issa Bhaloo, S.; Wu, Y.; LE Bras, A.; Yu, B.; Gu, W.; Xie, Y.; Deng, J.; Wang, Z.; Zhang, Z.; Kong, D.; et al. Binding of Dickkopf-3 to CXCR7 Enhances Vascular Progenitor Cell Migration and Degradable Graft Regeneration. Circ. Res. 2018, 123, 451–466. [Google Scholar] [CrossRef]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shiozawa, Y.; Wang, J.; Wang, Y.; Jung, Y.; Pienta, K.J.; Mehra, R.; Loberg, R.; Taichman, R.S. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 2008, 283, 4283–4294. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, S.; Mino, N.; Takahashi, T.; Sonobe, M.; Nagai, S.; Okubo, K.; Wada, H.; Date, H.; Miyahara, R. Higher expression of chemokine receptor CXCR7 is linked to early and metastatic recurrence in pathological stage I nonsmall cell lung cancer. Cancer 2009, 115, 2580–2593. [Google Scholar] [CrossRef]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 144. [Google Scholar] [CrossRef]
- Wani, N.A.; Nasser, M.W.; Ahirwar, D.K.; Zhao, H.; Miao, Z.; Shilo, K.; Ganju, R.K. C-X-C motif chemokine 12/C-X-C chemokine receptor type 7 signaling regulates breast cancer growth and metastasis by modulating the tumor microenvironment. Breast Cancer Res. 2014, 16, R54. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Han, M.-M.; Wang, F.; Xu, L.-L.; Yu, H.-X.; Yang, P.-Y. CXCR7 stimulates MAPK signaling to regulate hepatocellular carcinoma progression. Cell Death Dis. 2014, 5, e1488. [Google Scholar] [CrossRef] [Green Version]
- Salazar, N.; Muñoz, D.; Kallifatidis, G.; Singh, R.K.; Jordà, M.; Lokeshwar, B.L. The chemokine receptor CXCR7 interacts with EGFR to promote breast cancer cell proliferation. Mol. Cancer 2014, 13, 198. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Lokeshwar, B.L. The IL-8 regulated Chemokine Receptor CXCR7 Stimulates EGFR Signaling to Promote Prostate Cancer Growth. Cancer Res. 2011, 71, 3268–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Li, X.; Qian, X.; Wang, Y.; Lee, J.-H.; Xia, Y.; Hawke, D.H.; Zhang, G.; Lyu, J.; Lu, Z. Secreted and O-GlcNAcylated MIF binds to the human EGF receptor and inhibits its activation. Nat. Cell Biol. 2015, 17, 1348–1355. [Google Scholar] [CrossRef] [Green Version]
- Kiessling, M.K.; Schuierer, S.; Stertz, S.; Beibel, M.; Bergling, S.; Knehr, J.; Carbone, W.; de Vallière, C.; Tchinda, J.; Bouwmeester, T.; et al. Identification of oncogenic driver mutations by genome-wide CRISPR-Cas9 dropout screening. BMC Genom. 2016, 17, 723. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal Growth Factor Receptor (EGFR) Signaling Regulates Global Metabolic Pathways in EGFR-mutated Lung Adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.K.; Choi, Y.J.; Ryoo, B.-Y.; Na, I.I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. p53 enhances gefitinib-induced growth inhibition and apoptosis by regulation of Fas in non-small cell lung cancer. Cancer Res. 2007, 67, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.-T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar] [CrossRef]
- Bremer, E.; van Dam, G.M.; de Bruyn, M.; van Riezen, M.; Dijkstra, M.; Kamps, G.; Helfrich, W.; Haisma, H. Potent Systemic Anticancer Activity of Adenovirally Expressed EGFR-Selective TRAIL Fusion Protein. Mol. Ther. 2018, 16, 1919–1926. [Google Scholar] [CrossRef]
- Maussang, D.; Mujić-Delić, A.; Descamps, F.J.; Stortelers, C.; Vanlandschoot, P.; Stigter-van Walsum, M.; Vischer, H.F.; van Roy, M.; Vosjan, M.; Gonzalez-Pajuelo, M.; et al. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 2013, 288, 29562–29572. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Hyjek, E.; Vazquez, M.F.; Meherally, D.; Liu, Y.F.; Chadwick, P.A.; Rengifo, T.; Sica, G.L.; Port, J.L.; Lee, P.C.; et al. CXCL12 and CXCR4 in adenocarcinoma of the lung: Association with metastasis and survival. J. Thorac. Cardiovasc. Surg. 2009, 137, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, S. CXCR4/CXCL12 in Non-Small-Cell Lung Cancer Metastasis to the Brain. Int. J. Mol. Sci. 2013, 14, 1713–1727. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, M.; Hirsch, F.R. Clinical and comparative utility of afatinib in non-small cell lung cancer. Biologics 2014, 8, 183–192. [Google Scholar] [PubMed] [Green Version]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib restrains K-RAS–driven lung tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef] [PubMed]
- Wald, O.; Shapira, O.M.; Izhar, U. CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic roles and therapeutic potential. Theranostics 2013, 3, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Hisao, I.; Sunaga, N.; Shimizu, Y.; Yanagitani, N.; Kaira, K.; Tomizawa, Y.; Mori, M. SDF-1/CXCL12 overexpression and its role in the development of lung cancer. Cancer Res. 2008, 68, 5104. [Google Scholar]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, E.L.; Lee, W.; Lu, J.; Lowy, A.M.; Kim, J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med. 2012, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Borst, O.; Walker, B.; Fotinos, A.; Vogel, S.; Seizer, P.; Mack, A.; Alampour-Rajabi, S.; Rath, D.; Geisler, T.; et al. Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ. Res. 2014, 115, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Tripathi, V.; Ahmad, M.; Nath, N.; Mir, R.A.; Chauhan, S.S.; Luthra, K. CXCR7 mediated Giα independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell. Immunol. 2012, 272, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixido, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzon, M.; Codony-Servat, C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef]
- Tarnowski, M.; Kucia, M.; Ratajczak, M.Z. Isolation and Functional Analysis of CXCR7 Promoter—A Novel Receptor for Stromal Derived Factor-1 (SDF-1): Different Regulation of Expression in Human Hematopoietic Cells Versus Pediatric Sarcomas. Blood 2009, 114, 4583. [Google Scholar]
- Shostak, K.; Chariot, A. EGFR and NF-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Galvani, E.; Sun, J.; Leon, L.G.; Sciarrillo, R.; Narayan, R.S.; Sjin, R.T.T.; Lee, K.; Ohashi, K.; Heideman, D.A.M.; Alfieri, R.R.; et al. NF-kappaB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget 2015, 6, 42717–42732. [Google Scholar] [CrossRef] [PubMed]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-κB activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Okamoto, I.; Okamoto, W.; Tanaka, K.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K. Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations. Cancer Res. 2010, 70, 10402–10410. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kawasaki, N.; Taguchi, M.; Kabasawa, K. Survival impact of epidermal growth factor receptor overexpression in patients with non-small cell lung cancer: A meta-analysis. Thorax 2006, 61, 140–145. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination Treatment | TKIs (µM) | CXCR7i (µM) | FA | CI | Effect |
|---|---|---|---|---|---|
| Afatinib + CXCR7i | 10 | 10 | 0.84 | 0.47 | Synergistic |
| Erlotinib + CXCR7i | 10 | 10 | 0.55 | 0.74 | Moderately Synergistic |
| Gefitinib + CXCR7i | 10 | 10 | 0.47 | >1.1 | Antagonistic |
| Subtype | Strong (%) | Weak (%) | Strong-Weak (%) | Total | p-Value |
|---|---|---|---|---|---|
| Histology | 0.005 * | ||||
| ADC | 23 (92) | 2 (8) | 0 | 25 | |
| SQCC | 8 (44.5) | 6 (33.3) | 4 (22.2) | 18 | |
| Other | 4 (100) | 0 | 0 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Song, S.; Setroikromo, R.; Chen, S.; Hu, W.; Chen, D.; van der Wekken, A.J.; Melgert, B.N.; Timens, W.; van den Berg, A.; et al. CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor. Cancers 2019, 11, 455. https://doi.org/10.3390/cancers11040455
Liu B, Song S, Setroikromo R, Chen S, Hu W, Chen D, van der Wekken AJ, Melgert BN, Timens W, van den Berg A, et al. CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor. Cancers. 2019; 11(4):455. https://doi.org/10.3390/cancers11040455
Chicago/Turabian StyleLiu, Bin, Shanshan Song, Rita Setroikromo, Siwei Chen, Wenteng Hu, Deng Chen, Anthonie J. van der Wekken, Barbro N. Melgert, Wim Timens, Anke van den Berg, and et al. 2019. "CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor" Cancers 11, no. 4: 455. https://doi.org/10.3390/cancers11040455
APA StyleLiu, B., Song, S., Setroikromo, R., Chen, S., Hu, W., Chen, D., van der Wekken, A. J., Melgert, B. N., Timens, W., van den Berg, A., Saber, A., & Haisma, H. J. (2019). CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor. Cancers, 11(4), 455. https://doi.org/10.3390/cancers11040455