Ion Channels: New Actors Playing in Chemotherapeutic Resistance

 , , , ,
, , , ,  and
and
Abstract
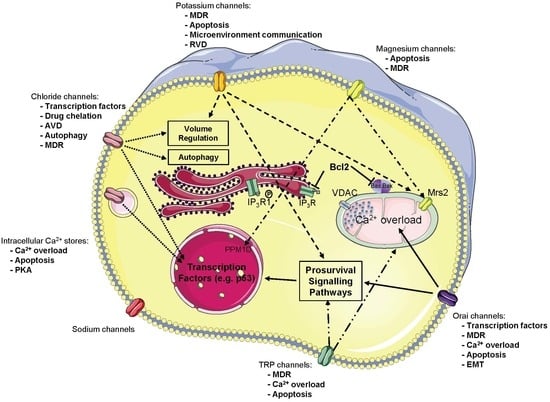
{kind=link}
{kind=link}
{kind=link}
{kind=link}
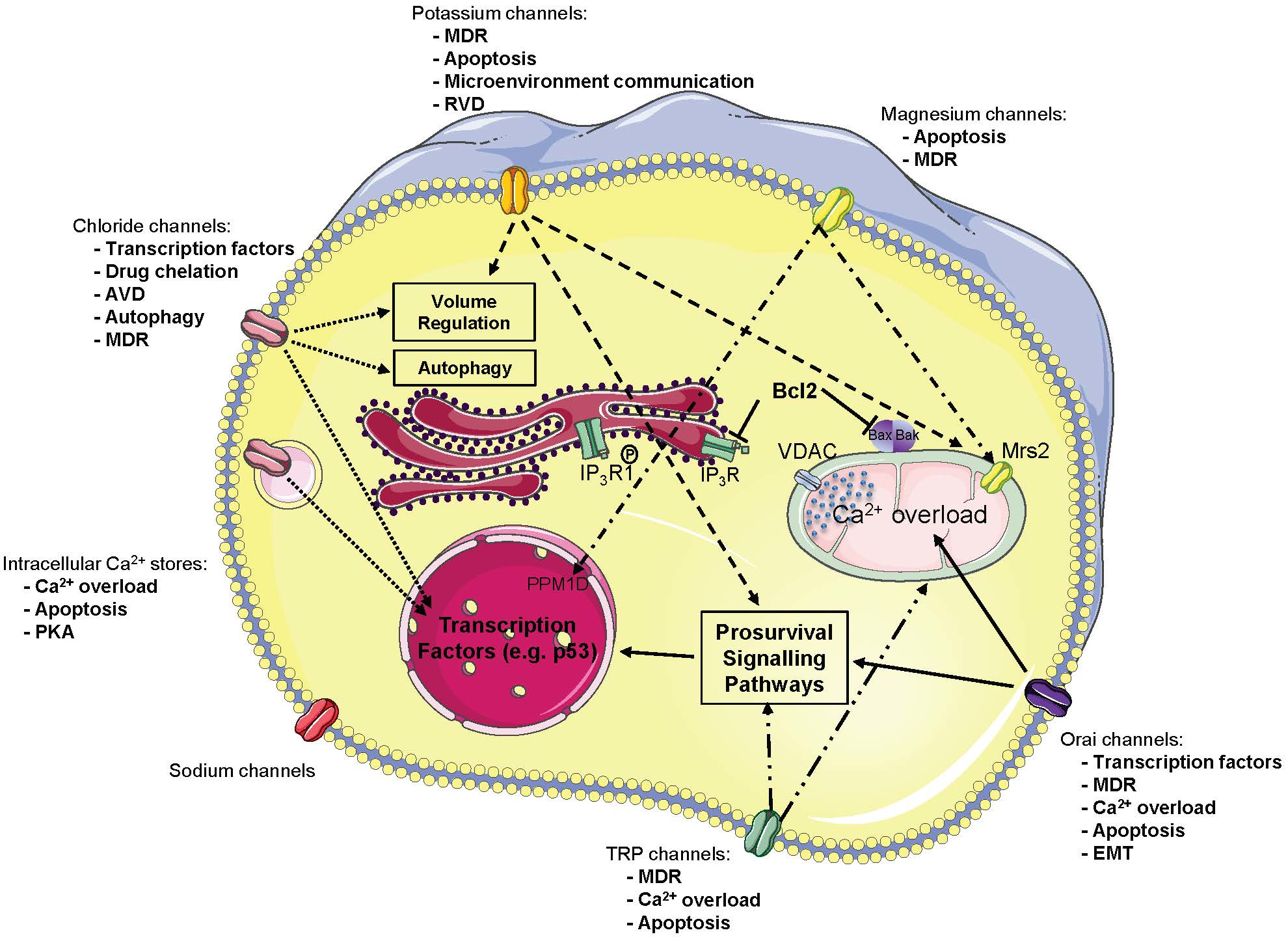
{kind=link}
{kind=link}
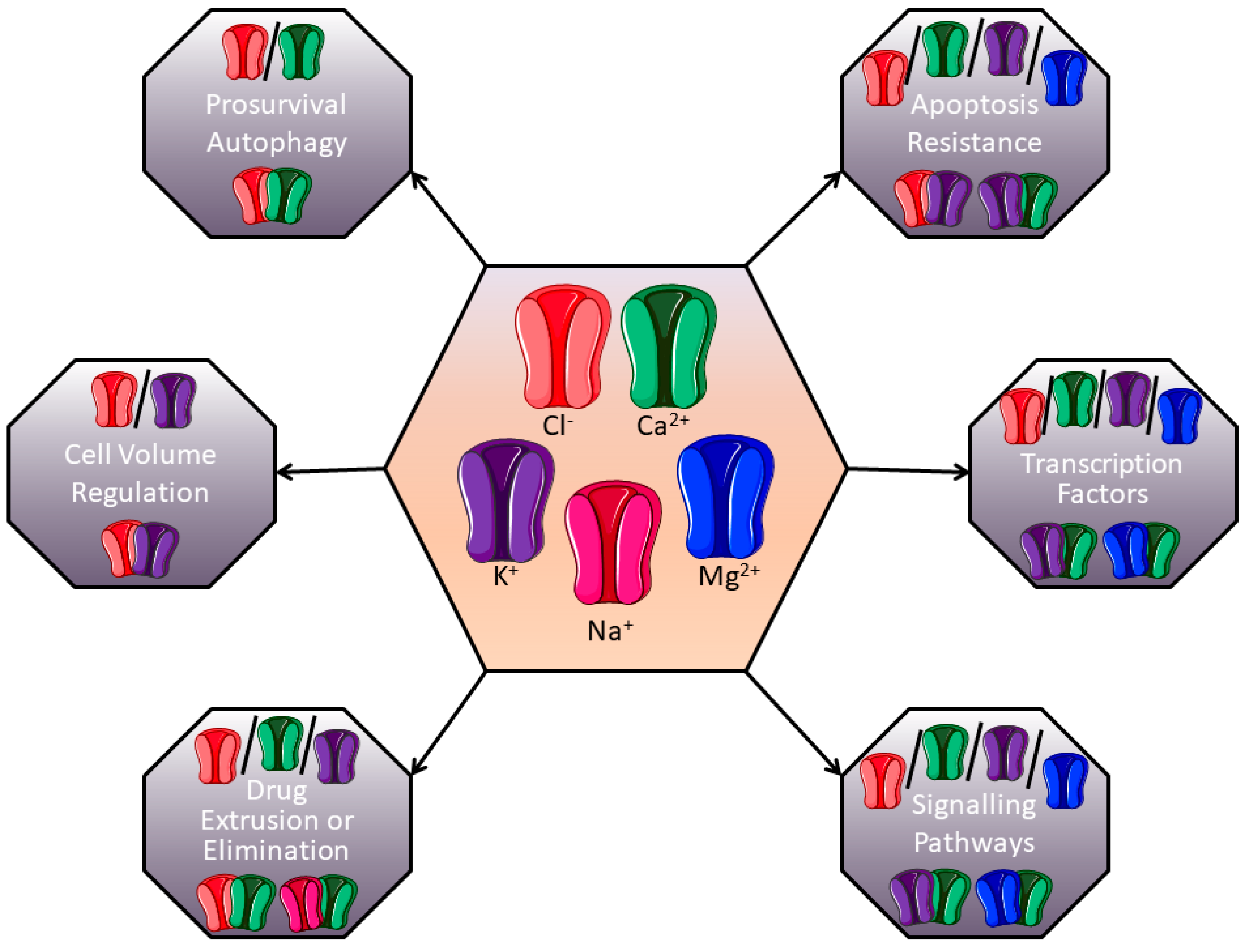{kind=link}
1. Introduction
- -
- Decreased activity of the uptake transporters, or alternatively enhanced efflux for water-soluble drugs (such as cisplatin)
- -
- Increased drug efflux mediated by energy-dependent transporters. For hydrophobic drugs (such as vinblastine, doxorubicin, and paclitaxel), entry occurs largely by diffusion across the membrane, although this process can also be critically enhanced by transport proteins
- -
- Indirect mechanisms by which transporters and channels affect chemosensitivity by modulating apoptosis pathways or the efficiency of drug diffusion along electrochemical gradients into cells
2. Calcium Channels
2.1. Plasma Membrane Ca2+ Channels
- -
- Voltage-operated channels, activated by membrane depolarization
- -
- Second messenger-operated channels, activated by small messenger molecules such as inositol phosphates, cyclic nucleotides, and lipid-derived messengers (diacyl-glycerol, arachidonic acid and its metabolites)
- -
- Receptor-operated channels, activated by direct binding of a neurotransmitter or hormone agonist
- -
- Store-operated channels (SOC), activated by depletion of intracellular Ca2+ stores [14]
2.1.1. Orai/Stim Mediated SOCE and Chemoresistance
Increased Resistance Conferred by the Overexpression of Stim and Orai Proteins
Reduced Resistance Conferred by the Overexpression of Stim and Orai Proteins
2.1.2. Transient Receptor Potential (TRP) Channels
2.1.3. Voltage-Gated Calcium Channels and Chemotherapy
2.2. Intracellular Calcium Stores
2.2.1. Intracellular Ca2+ Channels Expression Is Associated to Chemoresistance
2.2.2. Chemotherapy Modulates Intracellular Ca2+ Channels Activity
2.2.3. Signalling Pathways Involved in Chemoresistance Related to Intracellular Ca2+ Channels
2.2.4. Intracellular Ca2+ Channels: Targets to Overcome Chemoresistance
3. Potassium Channels
3.1. Correlation between the Expression Levels of K+ Channels and Sensitivity to Chemotherapeutic Drugs
3.2. Chemotherapy Modulates K+ Channel Activity
3.3. Signalling Pathways Involved in Chemoresistance Related to K+ Channels
3.4. K+ Channels: Targets to Overcome Chemoresistance
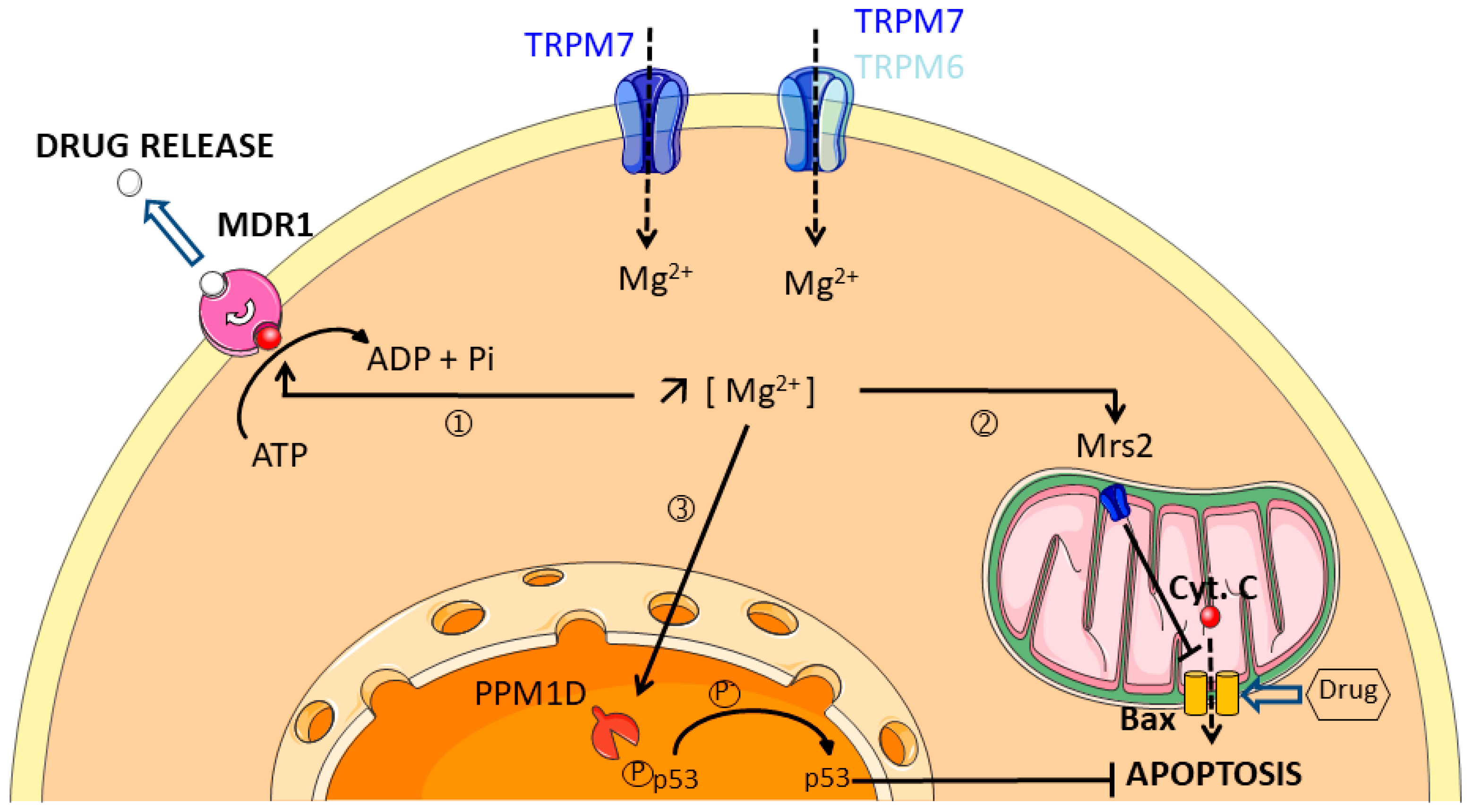
4. Magnesium Channels
5. Chloride Channels
5.1. Volume Regulated Chloride Channel Reduction and Promotion of Chemoresistance
5.2. Upregulation of Chloride Channels and Chemoresistance
6. Other Ion Channels
6.1. Voltage-Gated Sodium Channel in the Chemoresistance Process
6.2. ASIC Channels in the Chemoresistance Process
7. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-Inducing Factor |
| ALL | Acute Lymphoblastic Leukemia |
| ANO1 | ANOctamin 1 |
| ASIC | Acid-Sensing Ion Channel |
| AVD | Apoptosis Volume Decrease |
| Bax | Bcl-2-Associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BKCa | Large conductance, Ca2+-activated K+ channels |
| CFTR | Cystic Fibrosis Transmembrane conductance Regulator |
| ClC | Chloride Channel |
| CLIC1 | Chloride Intracellular Channel 1 |
| CXCR4 | C-X-C Chemokine Receptor 4 |
| ER | Endoplasmic Reticulum |
| EMT | Epithelial to Mesenchymal Transition |
| HCC | HepatoCellular Carcinoma |
| hEag1 | human Ether-à-Go-Go |
| hERG | human Ether-à-Go-Go-Related Gene |
| IP3R | Inositol 1,4,5-trisphosphate Receptor |
| MCU | Mitochondrial Calcium Uniport |
| MDR | MultiDrug Resistance |
| miRNA | micro-RNA |
| MMP | Matrix MetalloProteinase |
| MRP1 | Multidrug Resistance-associated Protein 1 |
| MSC | Bone Marrow Mesenchymal Cells |
| RVD | Regulatory Volume Decrease |
| SMAC | Second Mitochondria-derived Activator of Caspases |
| SK3 | Small conductance, Ca2+ activated K+ channel 3 |
| SOC channels | Store-Operated Calcium channels |
| SOCE | Store-Operated Calcium Entry |
| TAT-IDPs | TAT-fused inositol 1,4,5-trisphosphate receptor derived peptide |
| TRAIL | Tumor-necrosis-factor Related Apoptosis Inducing Ligand |
| VDAC | Voltage-Dependent Anion Channel |
| VRAC | Volume-Regulated Anion Channel |
References
- Gonzalez-Angulo, A.M.; Morales-Vasquez, F.; Hortobagyi, G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007, 608, 1–22. [Google Scholar]
- Li, S.; Kennedy, M.; Payne, S.; Kennedy, K.; Seewaldt, V.L.; Pizzo, S.V.; Bachelder, R.E. Model of Tumor Dormancy/Recurrence after Short-Term Chemotherapy. PLoS ONE 2014, 9, e98021. [Google Scholar] [CrossRef]
- Ghandadi, M.; Behravan, J.; Abnous, K.; Mosaffa, F. Reactive Oxygen Species Mediate TNF-a Cytotoxic Effects in the Multidrug-Resistant Breast Cancer Cell Line MCF-7/MX. Oncol. Res. Treat. 2016, 39, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sadée, W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Siesjö, B.K. Historical Overview: Calcium, Ischemia, and Death of Brain Cells. Ann. N. Y. Acad. Sci. 1988, 522, 638–661. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Anderle, P.; Bussey, K.J.; Barbacioru, C.; Shankavaram, U.; Dai, Z.; Reinhold, W.C.; Papp, A.; Weinstein, J.N.; Sadée, W. Membrane Transporters and Channels: Role of the Transportome in Cancer Chemosensitivity and Chemoresistance. Cancer Res. 2004, 64, 4294–4301. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Büsselberg, D.; Florea, A.-M. Targeting Intracellular Calcium Signaling ([Ca2+](i)) to Overcome Acquired Multidrug Resistance of Cancer Cells: A Mini-Overview. Cancers 2017, 9, 48. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W. Store-Operated Calcium Channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Capacitative calcium entry: From concept to molecules. Immunol. Rev. 2009, 231, 10–22. [Google Scholar] [CrossRef]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Abeele, F.V.; Skryma, R.; Shuba, Y.; Van Coppenolle, F.; Slomianny, C.; Roudbaraki, M.; Mauroy, B.; Wuytack, F.; Prevarskaya, N. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 2002, 1, 169–179. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Gueguinou, M.; Renaudineau, Y.; Shoji, K.F.; Felix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef]
- Helson, L. Calcium Channel Blocker Enhancement of Anticancer Drug Cytotoxicity—A Review. Cancer Drug Deliv. 1984, 1, 353–361. [Google Scholar] [CrossRef]
- Simpson, W.G. The calcium channel blocker verapamil and cancer chemotherapy. Cell Calcium 1985, 6, 449–467. [Google Scholar] [CrossRef]
- Mason, R.P. Effects of calcium channel blockers on cellular apoptosis. Cancer 1999, 85, 2093–2102. [Google Scholar] [CrossRef]
- Kiwit, J.C.; Hertel, A.; Matuschek, A.E. Reversal of chemoresistance in malignant gliomas by calcium antagonists: Correlation with the expression of multidrug-resistant p-glycoprotein. J. Neurosurg. 1994, 81, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Khadra, N.; Bresson-Bepoldin, L.; Penna, A.; Chaigne-Delalande, B.; Ségui, B.; Levade, T.; Vacher, A.-M.; Reiffers, J.; Ducret, T.; Moreau, J.-F.; et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) β2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19072–19077. [Google Scholar] [CrossRef] [PubMed]
- Limnander, A.; Depeille, P.; Freedman, T.S.; Liou, J.; Leitges, M.; Kurosaki, T.; Roose, J.P.; Weiss, A. STIM1, PKC-δ and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nat. Immunol. 2011, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P.; Lannon, W.A. Activation of ERK1/2 by Store-Operated Calcium Entry in Rat Parotid Acinar Cells. PLoS ONE 2013, 8, e72881. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Chiu, W.-T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-Independent Activation of Orai1 by SPCA2 in Mammary Tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef]
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Babaer, D.; Amara, S.; Ivy, M.; Zhao, Y.; Lammers, P.E.; Titze, J.M.; Tiriveedhi, V. High salt induces P-glycoprotein mediated treatment resistance in breast cancer cells through store operated calcium influx. Oncotarget 2018, 9, 25193–25205. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Honig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hao, J.; Zhang, Y.; Yang, Z.; Cao, Y.; Lu, W.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W.; et al. Orai1 mediates tumor-promoting store-operated Ca2+ entry in human gastrointestinal stromal tumors via c-KIT and the extracellular signal—Regulated kinase pathway. Tumor Biol. 2017, 39, 1010428317691426. [Google Scholar] [CrossRef]
- Shuttleworth, T.J. Orai3—The ‘exceptional’ Orai? J. Physiol. 2012, 590, 241–257. [Google Scholar] [CrossRef]
- Schindl, R.; Bergsmann, J.; Frischauf, I.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Groschner, K.; Romanin, C. 2-Aminoethoxydiphenyl Borate Alters Selectivity of Orai3 Channels by Increasing Their Pore Size. J. Biol. Chem. 2008, 283, 20261–20267. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.B.; Dhasarathy, A. The calcium channel proteins ORAI3 and STIM1 mediate TGF-beta induced Snai1 expression. Oncotarget 2018, 9, 29468–29483. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 752–760. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Vanoverberghe, K.; Vanden Abeele, F.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Prevarskaya, N. Oncogenic TRP channels. Adv. Exp. Med. Biol. 2011, 704, 929–945. [Google Scholar] [PubMed]
- Deveci, H.A.; Nazıroğlu, M.; Nur, G. 5-Fluorouracil-induced mitochondrial oxidative cytotoxicity and apoptosis are increased in MCF-7 human breast cancer cells by TRPV1 channel activation but not Hypericum perforatum treatment. Mol. Cell. Biochem. 2018, 439, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sanchez, S.; El Hiani, Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef] [PubMed]
- Koh, D.W.; Powell, D.P.; Blake, S.D.; Hoffman, J.L.; Hopkins, M.M.; Feng, X. Enhanced cytotoxicity in triple-negative and estrogen receptorpositive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep. 2015, 34, 1589–1598. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Meng, Z.; Cao, H.; Zhu, G.; Liu, T.; Wang, X. Knockdown of TRPM8 suppresses cancer malignancy and enhances epirubicin-induced apoptosis in human osteosarcoma cells. Int. J. Biol. Sci. 2013, 10, 90–102. [Google Scholar] [CrossRef]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2016, 48, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci. Rep. 2017, 7, 3158. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Pan, Q.; Jiang, L.; Chen, Z.; Zhang, F.; Liu, Y.; Xing, H.; Shi, M.; Li, J.; Li, X.; et al. Tumor endothelial expression of P-glycoprotein upon microvesicular transfer of TrpC5 derived from adriamycin-resistant breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ning, K.; Lu, T.X.; Sun, X.; Jin, L.; Qi, X.; Jin, J.; Hua, D. Increasing circulating exosomes-carrying TRPC5 predicts chemoresistance in metastatic breast cancer patients. Cancer Sci. 2017, 108, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef]
- Wang, T.; Ning, K.; Sun, X.; Zhang, C.; Jin, L.F.; Hua, D. Glycolysis is essential for chemoresistance induced by transient receptor potential channel C5 in colorectal cancer. BMC Cancer 2018, 18, 207. [Google Scholar] [CrossRef]
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q.; et al. Regulation of Multi-drug Resistance in hepatocellular carcinoma cells is TRPC6/Calcium Dependent. Sci. Rep. 2016, 6, 23269. [Google Scholar] [CrossRef] [PubMed]
- Madan, E.; Gogna, R.; Keppler, B.; Pati, U. p53 Increases Intra-Cellular Calcium Release by Transcriptional Regulation of Calcium Channel TRPC6 in GaQ3-Treated Cancer Cells. PLoS ONE 2013, 8, e71016. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, S.; Zhao, W.; Duan, J.; Wang, Z.; Chen, H.; Tian, Y.; Wang, D.; Zhao, J.; An, T.; et al. Mechanistic Exploration of Cancer Stem Cell Marker Voltage-Dependent Calcium Channel alpha2delta1 Subunit-mediated Chemotherapy Resistance in Small-Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2148–2158. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-Type Ca2+ Channel Inhibition Sensitizes Ovarian Cancer to Carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Akl, H.; Bultynck, G. Altered Ca2+ signaling in cancer cells: Proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim. Biophys. Acta 2013, 1835, 180–193. [Google Scholar] [CrossRef]
- Giorgi, C.; De Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef]
- Tsunoda, T.; Koga, H.; Yokomizo, A.; Tatsugami, K.; Eto, M.; Inokuchi, J.; Hirata, A.; Masuda, K.; Okumura, K.; Naito, S. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2005, 24, 1396–1402. [Google Scholar] [CrossRef]
- Schrodl, K.; Oelmez, H.; Edelmann, M.; Huber, R.M.; Bergner, A. Altered Ca2+-homeostasis of cisplatin-treated and low level resistant non-small-cell and small-cell lung cancer cells. Cell Oncol. 2009, 31, 301–315. [Google Scholar] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H.; Levine, B.; Boise, L.H.; Thompson, C.B.; Hardwick, J.M. Bax-independent inhibition of apoptosis by Bcl-XL. Nature 1996, 379, 554–556. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar] [PubMed]
- Padar, S.; van Breemen, C.; Thomas, D.W.; Uchizono, J.A.; Livesey, J.C.; Rahimian, R. Differential regulation of calcium homeostasis in adenocarcinoma cell line A549 and its Taxol-resistant subclone. Br. J. Pharm. 2004, 142, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 334. [Google Scholar] [CrossRef]
- Huang, Z.; Lei, X.; Zhong, M.; Zhu, B.; Tang, S.; Liao, D. Bcl-2 small interfering RNA sensitizes cisplatin-resistant human lung adenocarcinoma A549/DDP cell to cisplatin and diallyl disulfide. Acta Biochim. Biophys. Sin. 2007, 39, 835–843. [Google Scholar] [CrossRef]
- Schaaf, A.; Sagi, S.; Langbein, S.; Trojan, L.; Alken, P.; Michel, M.S. Cytotoxicity of cisplatin in bladder cancer is significantly enhanced by application of bcl-2 antisense oligonucleotides. Urol. Oncol. 2004, 22, 188–192. [Google Scholar] [CrossRef]
- Xie, Q.; Su, J.; Jiao, B.; Shen, L.; Ma, L.; Qu, X.; Yu, C.; Jiang, X.; Xu, Y.; Sun, L. ABT737 reverses cisplatin resistance by regulating ER-mitochondria Ca2+ signal transduction in human ovarian cancer cells. Int. J. Oncol. 2016, 49, 2507–2519. [Google Scholar] [CrossRef]
- Zhang, K.; Heidrich, F.M.; DeGray, B.; Boehmerle, W.; Ehrlich, B.E. Paclitaxel accelerates spontaneous calcium oscillations in cardiomyocytes by interacting with NCS-1 and the InsP3R. J. Mol. Cell. Cardiol. 2010, 49, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.B.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 2015, 57, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Lin, K.J.; Yang, W.L.; Kao, Y.W.; Chen, T.W.; Chao, S.C.; Chang, P.M.; Liu, C.Y.; Tzeng, C.H.; Chao, Y.; et al. Gene expression-based chemical genomics identifies heat-shock protein 90 inhibitors as potential therapeutic drugs in cholangiocarcinoma. Cancer 2013, 119, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Murren, J.R.; Durivage, H.J.; Buzaid, A.C.; Reiss, M.; Flynn, S.D.; Carter, D.; Hait, W.N. Trifluoperazine as a modulator of multidrug resistance in refractory breast cancer. Cancer Chemother. Pharm. 1996, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Schleuning, M.; Brumme, V.; Wilmanns, W. Growth inhibition of human leukemic cell lines by the phenothiazine derivative fluphenazine. Anticancer Res. 1993, 13, 599–602. [Google Scholar]
- Yeh, C.T.; Wu, A.T.; Chang, P.M.; Chen, K.Y.; Yang, C.N.; Yang, S.C.; Ho, C.C.; Chen, C.C.; Kuo, Y.L.; Lee, P.Y.; et al. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am. J. Respir. Crit. Care Med. 2012, 186, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer 2017, 16, 217–227. [Google Scholar] [CrossRef]
- Xie, Q.; Xu, Y.; Gao, W.; Zhang, Y.; Su, J.; Liu, Y.; Guo, Y.; Dou, M.; Hu, K.; Sun, L. TAT-fused IP3R-derived peptide enhances cisplatin sensitivity of ovarian cancer cells by increasing ER Ca2+ release. Int. J. Mol. Med. 2017, 41, 809–817. [Google Scholar] [CrossRef]
- Xu, L.; Xie, Q.; Qi, L.; Wang, C.; Xu, N.; Liu, W.; Yu, Y.; Li, S.; Xu, Y. Bcl-2 overexpression reduces cisplatin cytotoxicity by decreasing ER-mitochondrial Ca2+ signaling in SKOV3 cells. Oncol. Rep. 2018, 39, 985–992. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef]
- Vo, T.T.; Ryan, J.; Carrasco, R.; Neuberg, D.; Rossi, D.J.; Stone, R.M.; Deangelo, D.J.; Frattini, M.G.; Letai, A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012, 151, 344–355. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.T.; Ryan, J.A.; Tammareddi, A.; Moore Vdel, G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.T.; et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]
- Davids, M.S.; Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012, 30, 3127–3135. [Google Scholar] [CrossRef]
- Attali, B.; Chandy, K.G.; Giese, M.H.; Grissmer, S.; Gutman, G.A.; Jan, L.Y.; Lazdunski, M.; Mckinnon, D.; Nerbonne, J.; Pardo, L.A.; et al. Voltage-Gated Potassium Channels. Available online: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=81 (accessed on 1 February 2019).
- Ouadid-Ahidouch, H.; Ahidouch, A.; Pardo, L.A. Kv10.1 K+ channel: From physiology to cancer. Pflügers Arch. Eur. J. Physiol. 2016, 468, 751–762. [Google Scholar] [CrossRef]
- Comes, N.; Serrano-Albarrás, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; Ramón y Cajal, S.; Ferreres, J.C.; Felipe, A. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2477–2492. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Gasparoli, L.; Arcangeli, A. Potassium channels: Novel emerging biomarkers and targets for therapy in cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 53–65. [Google Scholar] [CrossRef]
- Sharp, S.Y.; Mistry, P.; Valenti, M.R.; Bryant, A.P.; Kelland, L.R. Selective potentiation of platinum drug cytotoxicity in cisplatin-sensitive and -resistant human ovarian carcinoma cell lines by amphotericin B. Cancer Chemother. Pharmacol. 1994, 35, 137–143. [Google Scholar] [CrossRef]
- Liang, X.-J.; Yin, J.-J.; Zhou, J.-W.; Wang, P.C.; Taylor, B.; Cardarelli, C.; Kozar, M.; Forte, R.; Aszalos, A.; Gottesman, M.M. Changes in biophysical parameters of plasma membranes influence cisplatin resistance of sensitive and resistant epidermal carcinoma cells. Exp. Cell Res. 2004, 293, 283–291. [Google Scholar] [CrossRef]
- Marklund, L.; Andersson, B.; Behnam-Motlagh, P.; Sandstrom, P.E.; Henriksson, R.; Grankvist, K. Cellular potassium ion deprivation enhances apoptosis induced by cisplatin. Basic Clin. Pharmacol. Toxicol. 2004, 94, 245–251. [Google Scholar] [CrossRef]
- Marklund, L.; Henriksson, R.; Grankvist, K. Cisplatin-induced apoptosis of mesothelioma cells is affected by potassium ion flux modulator amphotericin B and bumetanide. Int. J. Cancer 2001, 93, 577–583. [Google Scholar] [CrossRef]
- Liu, X.; Wei, L.; Zhao, B.; Cai, X.; Dong, C.; Yin, F. Low expression of KCNN3 may affect drug resistance in ovarian cancer. Mol. Med. Rep. 2018, 18, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.; Pink, R.C.; Caley, D.P.; Currie, J.M.; Brooks, S.A.; Carter, D.R. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 2565–2573. [Google Scholar] [CrossRef]
- Chen, S.Z.; Jiang, M.; Zhen, Y.S. HERG K+ channel expression-related chemosensitivity in cancer cells and its modulation by erythromycin. Cancer Chemother. Pharmacol. 2005, 56, 212–220. [Google Scholar] [CrossRef]
- Han, Y.; Shi, Y.; Han, Z.; Sun, L.; Fan, D. Detection of potassium currents and regulation of multidrug resistance by potassium channels in human gastric cancer cells. Cell Biol. Int. 2007, 31, 741–747. [Google Scholar] [CrossRef]
- Leanza, L.; O’Reilly, P.; Doyle, A.; Venturini, E.; Zoratti, M.; Szegezdi, E.; Szabo, I. Correlation between potassium channel expression and sensitivity to drug-induced cell death in tumor cell lines. Curr. Pharm. Des. 2014, 20, 189–200. [Google Scholar] [CrossRef]
- Lam, H.D.; Lemay, A.M.; Kelly, J.; Hill, C.E. Loss of Kv and MaxiK currents associated with increased MRP1 expression in small cell lung carcinoma. J. Cell. Physiol. 2006, 209, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.J.; Taylor, B.; Cardarelli, C.; Yin, J.J.; Annereau, J.P.; Garfield, S.; Wincovitch, S.; Szakacs, G.; Gottesman, M.M.; Aszalos, A. Different roles for K+ channels in cisplatin-resistant cell lines argue against a critical role for these channels in cisplatin resistance. Anticancer Res. 2005, 25, 4113–4122. [Google Scholar] [PubMed]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef]
- Agarwal, J.R.; Griesinger, F.; Stuhmer, W.; Pardo, L.A. The potassium channel Ether a go-go is a novel prognostic factor with functional relevance in acute myeloid leukemia. Mol. Cancer 2010, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.; Lan, Z.; Yue-li, L.; Li-lin, H. Knockdown of Eag1 Expression by RNA Interference Increases Chemosensitivity to Cisplatin in Ovarian Cancer Cells. Reprod. Sci. 2015, 22, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Hasegawa, Y.; Shimizu, T.; Okada, Y. IK1 channel activity contributes to cisplatin sensitivity of human epidermoid cancer cells. Am. J. Physiol. Cell Physiol. 2008, 294, C1398–C1406. [Google Scholar] [CrossRef]
- Splettstoesser, F.; Florea, A.M.; Busselberg, D. IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Jirsch, J.; Deeley, R.G.; Cole, S.P.; Stewart, A.J.; Fedida, D. Inwardly rectifying K+ channels and volume-regulated anion channels in multidrug-resistant small cell lung cancer cells. Cancer Res. 1993, 53, 4156–4160. [Google Scholar]
- Huang, M.H.; Huang, Y.M.; Wu, S.N. The Inhibition by Oxaliplatin, a Platinum-Based Anti-Neoplastic Agent, of the Activity of Intermediate-Conductance Ca2+-Activated K+ Channels in Human Glioma Cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 1390–1406. [Google Scholar] [CrossRef]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. K+ channels in apoptosis. J. Membr. Biol. 2006, 209, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Azzolini, M.; Peruzzo, R.; Capitanio, P.; Leanza, L. Mitochondrial potassium channels in cell death. Biochem. Biophys. Res. Commun. 2018, 500, 51–58. [Google Scholar] [CrossRef]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef] [PubMed]
- Quast, S.A.; Berger, A.; Buttstadt, N.; Friebel, K.; Schonherr, R.; Eberle, J. General Sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS ONE 2012, 7, e39290. [Google Scholar] [CrossRef]
- Zhang, R.; Tian, P.; Chi, Q.; Wang, J.; Wang, Y.; Sun, L.; Liu, Y.; Tian, S.; Zhang, Q. Human ether-a-go-go-related gene expression is essential for cisplatin to induce apoptosis in human gastric cancer. Oncol. Rep. 2012, 27, 433–440. [Google Scholar]
- Choi, S.Y.; Kim, H.R.; Ryu, P.D.; Lee, S.Y. Regulation of voltage-gated potassium channels attenuates resistance of side-population cells to gefitinib in the human lung cancer cell line NCI-H460. BMC Pharmacol. Toxicol. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, J.; Peng, J.; Wu, X.; Zhang, Y.; Zhu, W.; Guo, L. Upregulation of the inwardly rectifying potassium channel Kir2.1 (KCNJ2) modulates multidrug resistance of small-cell lung cancer under the regulation of miR-7 and the Ras/MAPK pathway. Mol. Cancer 2015, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-a-go-go (EAG1). Eur. J. Cancer 2013, 49, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Masselli, M.; De Lorenzo, E.; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D’Amico, M.; Basso, G.; et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood 2011, 117, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Lopez, M.G.; Zuniga-Garcia, V.; Hernandez-Gallegos, E.; Vera, E.; Chasiquiza-Anchatuna, C.A.; Viteri-Yanez, M.; Sanchez-Ramos, J.; Garrido, E.; Camacho, J. The combination astemizole-gefitinib as a potential therapy for human lung cancer. Oncotargets Ther. 2017, 10, 5795–5803. [Google Scholar] [CrossRef]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef]
- de Baaij, J.H.; Hoenderop, J.G.; Bindels, R.J. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Dibaba, D.; Xun, P.; Yokota, K.; White, E.; He, K. Magnesium intake and incidence of pancreatic cancer: The VITamins and Lifestyle study. Br. J. Cancer 2015, 113, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shrubsole, M.J.; Ness, R.M.; Hibler, E.A.; Cai, Q.; Long, J.; Chen, Z.; Li, G.; Jiang, M.; Hou, L.; et al. Calcium/magnesium intake ratio, but not magnesium intake, interacts with genetic polymorphism in relation to colorectal neoplasia in a two-phase study. Mol. Carcinog. 2016, 55, 1449–1457. [Google Scholar] [CrossRef]
- Castiglioni, S.; Cazzaniga, A.; Trapani, V.; Cappadone, C.; Farruggia, G.; Merolle, L.; Wolf, F.I.; Iotti, S.; Maier, J.A. Magnesium homeostasis in colon carcinoma LoVo cells sensitive or resistant to doxorubicin. Sci. Rep. 2015, 5, 16538. [Google Scholar] [CrossRef]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 is required for breast tumor cell metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, X.; Yan, P.; Han, Y.; Sun, S.; Wu, K.; Fan, D. Human mitochondrial Mrs2 protein promotes multidrug resistance in gastric cancer cells by regulating p27, cyclin D1 expression and cytochrome C release. Cancer Biol. Ther. 2009, 8, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Hamada, H.; Tsuruo, T. ATP/Mg2+-dependent binding of vincristine to the plasma membrane of multidrug-resistant K562 cells. J. Biol. Chem. 1988, 263, 11887–11891. [Google Scholar] [PubMed]
- Hamada, H.; Tsuruo, T. Characterization of the ATPase activity of the Mr 170,000 to 180,000 membrane glycoprotein (P-glycoprotein) associated with multidrug resistance in K562/ADM cells. Cancer Res. 1988, 48, 4926–4932. [Google Scholar] [PubMed]
- Naito, M.; Tsuruo, T. Competitive inhibition by verapamil of ATP-dependent high affinity vincristine binding to the plasma membrane of multidrug-resistant K562 cells without calcium ion involvement. Cancer Res. 1989, 49, 1452–1455. [Google Scholar]
- Ali, A.Y.; Kim, J.Y.; Pelletier, J.F.; Vanderhyden, B.C.; Bachvarov, D.R.; Tsang, B.K. Akt confers cisplatin chemoresistance in human gynecological carcinoma cells by modulating PPM1D stability. Mol. Carcinog. 2015, 54, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713 e706. [Google Scholar] [CrossRef]
- Cao, Y.; Liao, C.; Tan, A.; Liu, L.; Gao, F. Meta-analysis of incidence and risk of hypomagnesemia with cetuximab for advanced cancer. Chemotherapy 2010, 56, 459–465. [Google Scholar] [CrossRef]
- Petrelli, F.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Barni, S. Risk of anti-EGFR monoclonal antibody-related hypomagnesemia: Systematic review and pooled analysis of randomized studies. Expert Opin. Drug Saf. 2012, 11 (Suppl. 1), S9–S19. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, L.; Li, H.; Liu, B.; Zou, Z. Incidence and risk of hypomagnesemia in advanced cancer patients treated with cetuximab: A meta-analysis. Oncol. Lett. 2013, 5, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qi, Y.; Zhang, D.; Gong, C.; Yao, A.; Xiao, Y.; Yang, J.; Zhou, F.; Zhou, Y. Electrolyte disorders assessment in solid tumor patients treated with anti-EGFR monoclonal antibodies: A pooled analysis of 25 randomized clinical trials. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.C.; Wu, C.F.; Chen, C.W.; Shi, C.S.; Huang, W.S.; Kuan, F.C. Hypomagnesemia and clinical benefits of anti-EGFR monoclonal antibodies in wild-type KRAS metastatic colorectal cancer: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 2047. [Google Scholar] [CrossRef]
- Duran, C.; Thompson, C.H.; Xiao, Q.; Hartzell, H.C. Chloride channels: Often enigmatic, rarely predictable. Annu. Rev. Physiol. 2010, 72, 95–121. [Google Scholar] [CrossRef]
- Peretti, M.; Angelini, M.; Savalli, N.; Florio, T.; Yuspa, S.H.; Mazzanti, M. Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1848, 2523–2531. [Google Scholar] [CrossRef]
- Wang, H.; Zou, L.; Ma, K.; Yu, J.; Wu, H.; Wei, M.; Xiao, Q. Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol. Cancer 2017, 16, 152. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Other ion channels. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S195–S207. [Google Scholar] [CrossRef]
- Marin, M.; Poret, A.; Maillet, G.; Leboulenger, F.; Le Foll, F. Regulation of volume-sensitive Cl- channels in multi-drug resistant MCF7 cells. Biochem. Biophys. Res. Commun. 2005, 334, 1266–1278. [Google Scholar] [CrossRef]
- Ise, T.; Shimizu, T.; Lee, E.L.; Inoue, H.; Kohno, K.; Okada, Y. Roles of volume-sensitive Cl- channel in cisplatin-induced apoptosis in human epidermoid cancer cells. J. Membr. Biol. 2005, 205, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Shimizu, T.; Ise, T.; Numata, T.; Kohno, K.; Okada, Y. Impaired activity of volume-sensitive Cl- channel is involved in cisplatin resistance of cancer cells. J. Cell. Physiol. 2007, 211, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 587, 2141–2149. [Google Scholar]
- Min, X.J.; Li, H.; Hou, S.C.; He, W.; Liu, J.; Hu, B.; Wang, J. Dysfunction of volume-sensitive chloride channels contributes to cisplatin resistance in human lung adenocarcinoma cells. Exp. Biol. Med. 2011, 236, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, K.A.; Andersen, E.C.; Hansen, C.F.; Klausen, T.K.; Hougaard, C.; Lambert, I.H.; Hoffmann, E.K. Deregulation of apoptotic volume decrease and ionic movements in multidrug-resistant tumor cells: Role of chloride channels. Am. J. Physiol. Cell Physiol. 2010, 298, C14–C25. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhu, L.; Lin, J.; Liu, S.; Luo, H.; Mao, J.; Nie, S.; Chen, L.; Wang, L. Cisplatin activates volume-sensitive like chloride channels via purinergic receptor pathways in nasopharyngeal carcinoma cells. J. Membr. Biol. 2015, 248, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Sorensen, B.H.; Sauter, D.P.; Lambert, I.H. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels 2015, 9, 380–396. [Google Scholar] [CrossRef]
- Shimizu, T.; Lee, E.L.; Ise, T.; Okada, Y. Volume-sensitive Cl− channel as a regulator of acquired cisplatin resistance. Anticancer Res. 2008, 28, 75–83. [Google Scholar]
- Voss, F.K.; Ullrich, F.; Munch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- Fujimoto, M.; Inoue, T.; Kito, H.; Niwa, S.; Suzuki, T.; Muraki, K.; Ohya, S. Transcriptional repression of HER2 by ANO1 Cl− channel inhibition in human breast cancer cells with resistance to trastuzumab. Biochem. Biophys. Res. Commun. 2017, 482, 188–194. [Google Scholar] [CrossRef]
- Kang, M.K.; Kang, S.K. Pharmacologic blockade of chloride channel synergistically enhances apoptosis of chemotherapeutic drug-resistant cancer stem cells. Biochem. Biophys. Res. Commun. 2008, 373, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Chen, Y.; Cao, G.; Liu, C.; Xu, J.; Deng, H.; Zhang, Z. Identification and validation of differentially expressed proteins in epithelial ovarian cancers using quantitative proteomics. Oncotarget 2016, 7, 83187–83199. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Cui, R.; Qu, H.; Liu, C.; Deng, H.; Zhang, Z. Expression and prognostic value of CLIC1 in epithelial ovarian cancer. Exp. Ther. Med. 2018, 15, 4943–4949. [Google Scholar] [CrossRef] [PubMed]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef]
- Weylandt, K.H.; Nebrig, M.; Jansen-Rosseck, N.; Amey, J.S.; Carmena, D.; Wiedenmann, B.; Higgins, C.F.; Sardini, A. ClC-3 expression enhances etoposide resistance by increasing acidification of the late endocytic compartment. Mol. Cancer Ther. 2007, 6, 979–986. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, H.; Kang, J.S.; Zhang, L.; Su, J.; Li, H.Y.; Sun, L.K. 5-Nitro-2-(3-phenylpropylamino) benzoic acid induced drug resistance to cisplatin in human erythroleukemia cell lines. Anat. Rec. 2011, 294, 945–952. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, L.; Zhang, J.; Zhang, L.; Yan, X.; Su, J. Suppression of chloride voltage-gated channel 3 expression increases sensitivity of human glioma U251 cells to cisplatin through lysosomal dysfunction. Oncol. Lett. 2018, 16, 835–842. [Google Scholar] [CrossRef]
- Fujimoto, M.; Kito, H.; Kajikuri, J.; Ohya, S. Transcriptional repression of human epidermal growth factor receptor 2 by ClC-3 Cl− /H+ transporter inhibition in human breast cancer cells. Cancer Sci. 2018, 109, 2781–2791. [Google Scholar] [CrossRef]
- Su, J.; Xu, Y.; Zhou, L.; Yu, H.M.; Kang, J.S.; Liu, N.; Quan, C.S.; Sun, L.K. Suppression of chloride channel 3 expression facilitates sensitivity of human glioma U251 cells to cisplatin through concomitant inhibition of Akt and autophagy. Anat. Rec. 2013, 296, 595–603. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, X.; Luo, Z.; Wang, S.; Lin, J.; Xie, Z.; Li, M.; Li, C.; Cao, H.; Huang, Q.; et al. Chloride channel-3 mediates multidrug resistance of cancer by upregulating P-glycoprotein expression. J. Cell. Physiol. 2018, 234, 6611–6623. [Google Scholar] [CrossRef]
- Zhang, H.; Pang, Y.; Ma, C.; Li, J.; Wang, H.; Shao, Z. ClC5 Decreases the Sensitivity of Multiple Myeloma Cells to Bortezomib via Promoting Prosurvival Autophagy. Oncol. Res. 2018, 26, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, D. CLIC1 Induces Drug Resistance in Human Choriocarcinoma Through Positive Regulation of MRP1. Oncol. Res. 2017, 25, 863–871. [Google Scholar] [CrossRef]
- House, C.D.; Wang, B.D.; Ceniccola, K.; Williams, R.; Simaan, M.; Olender, J.; Patel, V.; Baptista-Hon, D.T.; Annunziata, C.M.; Gutkind, J.S.; et al. Voltage-gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Sci. Rep. 2015, 5, 11541. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Yang, M.; Millican-Slater, R.; Brackenbury, W.J. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 2015, 6, 32914–32929. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.; Altun, S.; Gumushan, H.; Patel, A.; Djamgoz, M.B. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012, 323, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Gerard, V.; Rouzaire-Dubois, B.; Dilda, P.; Dubois, J.M. Alterations of ionic membrane permeabilities in multidrug-resistant neuroblastoma x glioma hybrid cells. J. Exp. Biol. 1998, 201, 21–31. [Google Scholar]
- Yamashita, N.; Hamada, H.; Tsuruo, T.; Ogata, E. Enhancement of voltage-gated Na+ channel current associated with multidrug resistance in human leukemia cells. Cancer Res. 1987, 47, 3736–3741. [Google Scholar]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway. Lab. Investig. A J. Tech. Methods Pathol. 2017, 97, 53–69. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1848, 2532–2546. [Google Scholar] [CrossRef]
- Kunzelmann, K. Ion channels in regulated cell death. Cell. Mol. Life Sci. 2016, 73, 2387–2403. [Google Scholar] [CrossRef]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; Van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2018, 9, 24653–24671. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Belpomme, D.; Gauthier, S.; Pujade-Lauraine, E.; Facchini, T.; Goudier, M.J.; Krakowski, I.; Netter-Pinon, G.; Frenay, M.; Gousset, C.; Marie, F.N.; et al. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann. Oncol. 2000, 11, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Lester-Coll, N.H.; Supko, J.G.; Kluytenaar, J.; Pavlik, K.F.; Yu, J.B.; Moliterno, J.; Piepmeier, J.; Becker, K.; Baehring, J.M.; Huttner, A.; et al. Mibefradil dihydrochoride with hypofractionated radiation for recurrent glioblastoma: A phase I dose expansion trial. J. Clin. Oncol. 2018, 36, e14046. [Google Scholar] [CrossRef]
- Yu, L.J.; Wall, B.A.; Chen, S. The current management of brain metastasis in melanoma: A focus on riluzole. Expert Rev. Neurother. 2015, 15, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kischel, P.; Girault, A.; Rodat-Despoix, L.; Chamlali, M.; Radoslavova, S.; Abou Daya, H.; Lefebvre, T.; Foulon, A.; Rybarczyk, P.; Hague, F.; et al. Ion Channels: New Actors Playing in Chemotherapeutic Resistance. Cancers 2019, 11, 376. https://doi.org/10.3390/cancers11030376
Kischel P, Girault A, Rodat-Despoix L, Chamlali M, Radoslavova S, Abou Daya H, Lefebvre T, Foulon A, Rybarczyk P, Hague F, et al. Ion Channels: New Actors Playing in Chemotherapeutic Resistance. Cancers. 2019; 11(3):376. https://doi.org/10.3390/cancers11030376
Chicago/Turabian StyleKischel, Philippe, Alban Girault, Lise Rodat-Despoix, Mohamed Chamlali, Silviya Radoslavova, Hiba Abou Daya, Thibaut Lefebvre, Arthur Foulon, Pierre Rybarczyk, Frédéric Hague, and et al. 2019. "Ion Channels: New Actors Playing in Chemotherapeutic Resistance" Cancers 11, no. 3: 376. https://doi.org/10.3390/cancers11030376
APA StyleKischel, P., Girault, A., Rodat-Despoix, L., Chamlali, M., Radoslavova, S., Abou Daya, H., Lefebvre, T., Foulon, A., Rybarczyk, P., Hague, F., Dhennin-Duthille, I., Gautier, M., & Ouadid-Ahidouch, H. (2019). Ion Channels: New Actors Playing in Chemotherapeutic Resistance. Cancers, 11(3), 376. https://doi.org/10.3390/cancers11030376