ADRB2-Targeting Therapies for Prostate Cancer

{kind=link}
{kind=link}
Abstract
1. Introduction
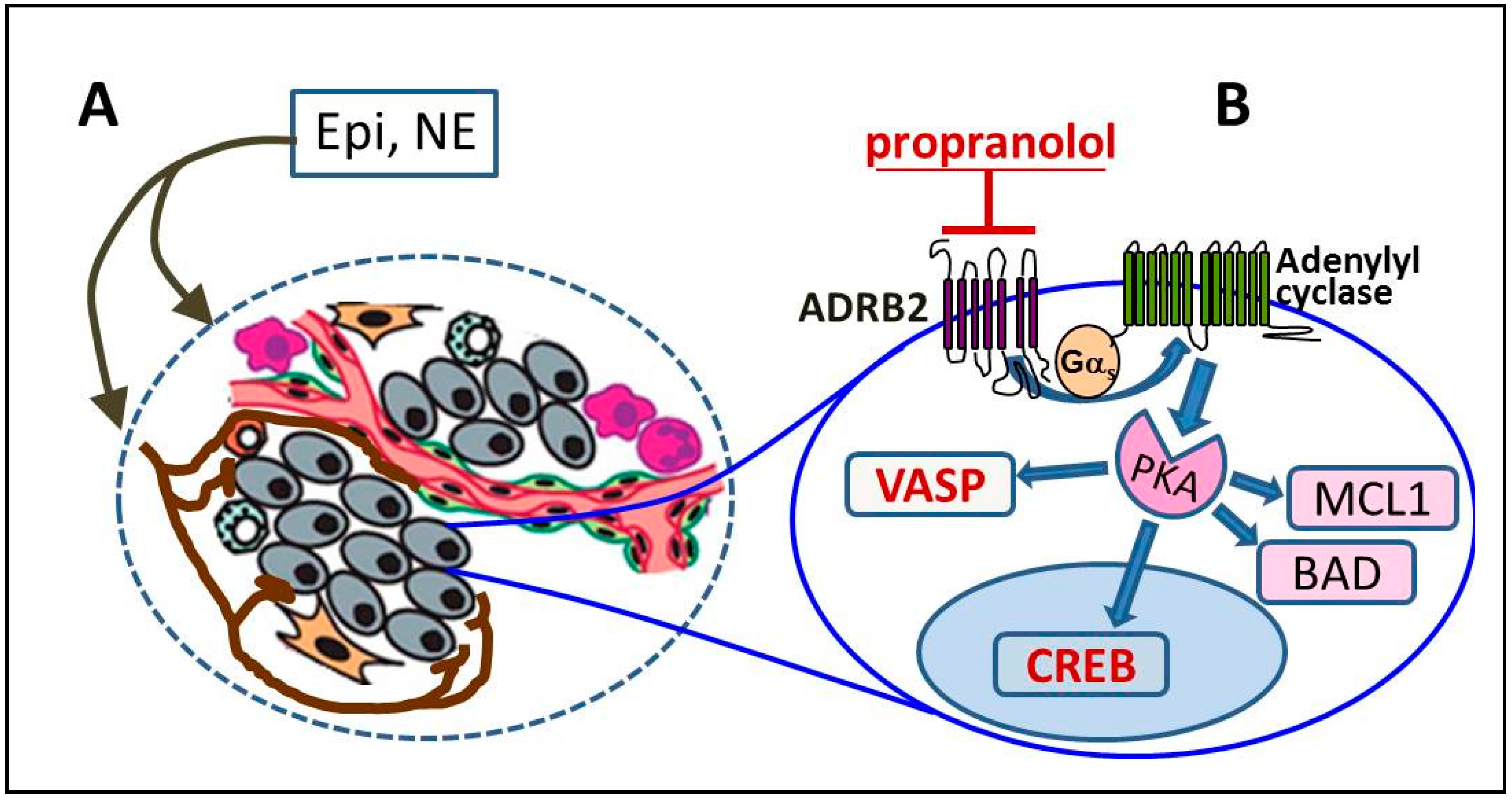
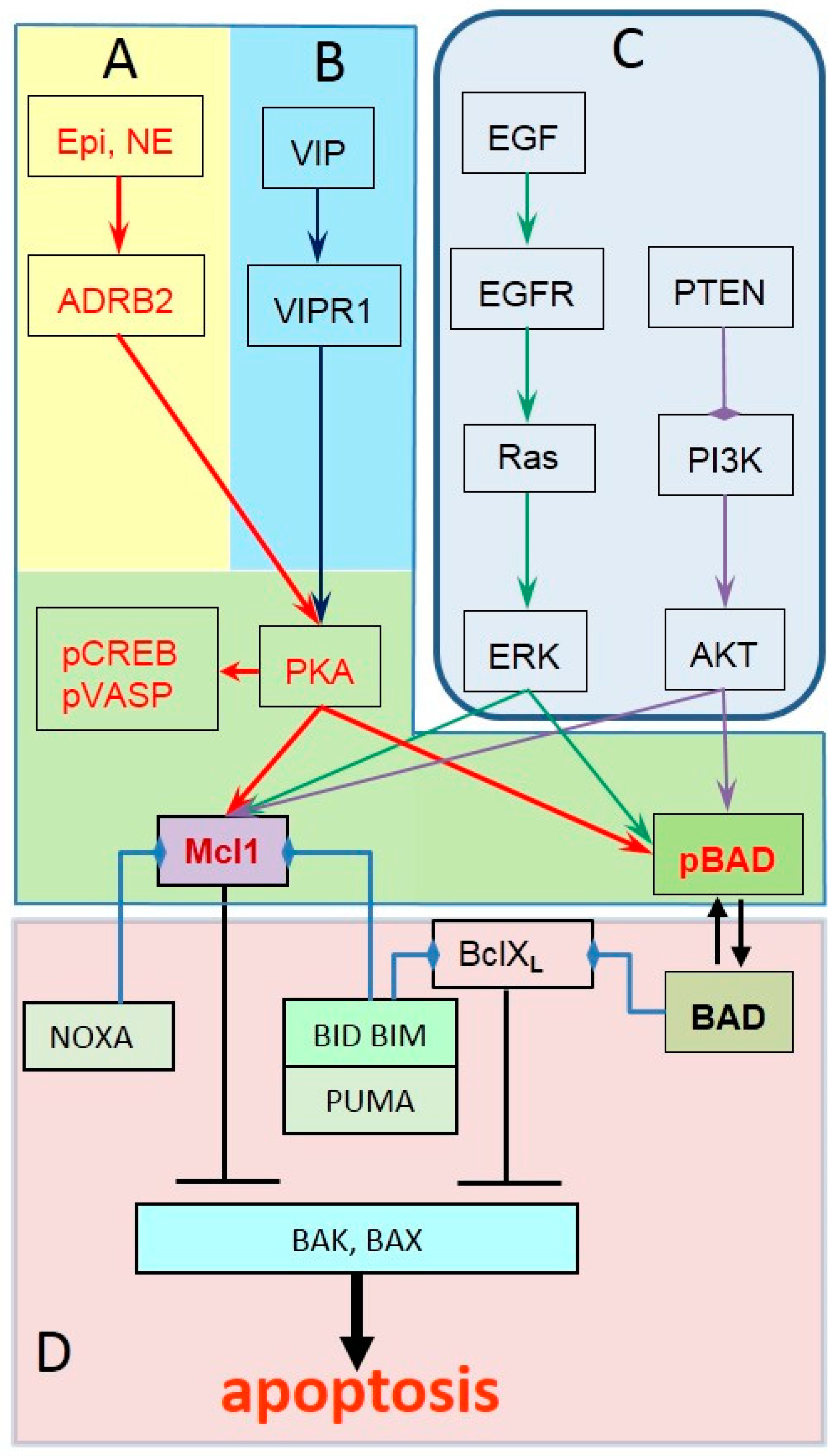
2. ADRB2 Signaling in Prostate Cancer Progression
3. Identifying Tumors with Active ADRB2 Signaling
4. Identifying Prostate Tumors Unresponsive to Propranolol
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; de Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef]
- Collins, D.C.; Sundar, R.; Lim, J.S.; Yap, T.A. Towards Precision Medicine in the Clinic: From Biomarker Discovery to Novel Therapeutics. Trends Pharmacol. Sci. 2017, 38, 25–40. [Google Scholar] [CrossRef]
- Singh, P.; Uzgare, A.; Litvinov, I.; Denmeade, S.R.; Isaacs, J.T. Combinatorial androgen receptor targeted therapy for prostate cancer. Endocr. Relat. Cancer 2006, 13, 653–666. [Google Scholar] [CrossRef]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. Beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2014, 4, 375. [Google Scholar] [CrossRef]
- Philipp, M.; Hein, L. Adrenergic receptor knockout mice: Distinct functions of 9 receptor subtypes. Pharmacol. Ther. 2004, 101, 65–74. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Gudermann, T.; Schoneberg, T.; Schultz, G. Functional and structural complexity of signal transduction via G-protein-coupled receptors. Annu. Rev. Neurosci. 1997, 20, 399–427. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta Physiol. (Oxf.) 2007, 190, 9–19. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Prichard, B.N.; Cruickshank, J.M.; Graham, B.R. Beta-Adrenergic blocking drugs in the treatment of hypertension. Blood Press 2001, 10, 366–386. [Google Scholar] [CrossRef]
- Baker, J.G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharm. 2005, 144, 317–322. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leitz, M.R.; Oberdorf-Maass, S.; Lohse, M.J.; Klotz, K.N. Comparative pharmacology of human beta-adrenergic receptor subtypes--characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedebergs Arch. Pharm. 2004, 369, 151–159. [Google Scholar] [CrossRef]
- Nagmani, R.; Pasco, D.S.; Salas, R.D.; Feller, D.R. Evaluation of beta-adrenergic receptor subtypes in the human prostate cancer cell line-LNCaP. Biochem. Pharm. 2003, 65, 1489–1494. [Google Scholar] [CrossRef]
- Poyet, P.; Gagne, B.; Lavoie, M.; Labrie, F. Characteristics of the beta-adrenergic receptor in the rat ventral prostate using [125I]cyanopindolol. Mol. Cell. Endocrinol. 1986, 48, 59–67. [Google Scholar] [CrossRef]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar] [CrossRef]
- Ramberg, H.; Eide, T.; Krobert, K.A.; Levy, F.O.; Dizeyi, N.; Bjartell, A.S.; Abrahamsson, P.A.; Tasken, K.A. Hormonal regulation of beta2-adrenergic receptor level in prostate cancer. Prostate 2008, 68, 1133–1142. [Google Scholar] [CrossRef]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Chen, A.J.; Sarma, J.V.; Zetoune, F.S.; McGuire, S.R.; List, R.P.; Day, D.E.; Hoesel, L.M.; et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 2007, 449, 721–725. [Google Scholar] [CrossRef]
- Marino, F.; Cosentino, M. Adrenergic modulation of immune cells: An update. Amino Acids 2013, 45, 55–71. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef]
- Palm, D.; Lang, K.; Niggemann, B.; Drell, T.L.; Masur, K.; Zaenker, K.S.; Entschladen, F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int. J. Cancer 2006, 118, 2744–2749. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R., Jr.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [CrossRef]
- Kulik, G. Personalized prostate cancer therapy based on systems analysis of the apoptosis regulatory network. Asian J. Androl. 2015, 17, 471–474. [Google Scholar]
- Sun, X.; Bao, J.; Nelson, K.C.; Li, K.C.; Kulik, G.; Zhou, X. Systems modeling of anti-apoptotic pathways in prostate cancer: Psychological stress triggers a synergism pattern switch in drug combination therapy. PLoS Comput. Biol. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Holmgreen, S.P.; Huang, D.C.; Adams, J.M.; Cory, S. Survival activity of Bcl-2 homologs Bcl-w and A1 only partially correlates with their ability to bind pro-apoptotic family members. Cell Death Differ. 1999, 6, 525–532. [Google Scholar] [CrossRef]
- Dutta, S.; Gulla, S.; Chen, T.S.; Fire, E.; Grant, R.A.; Keating, A.E. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 2010, 398, 747–762. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef]
- Ozaki, Y.; Kato, T.; Kitagawa, M.; Fujita, H.; Kitagawa, S. Calpain inhibition delays neutrophil apoptosis via cyclic AMP-independent activation of protein kinase A and protein kinase A-mediated stabilization of Mcl-1 and X-linked inhibitor of apoptosis (XIAP). Arch. Biochem. Biophys. 2008, 477, 227–231. [Google Scholar] [CrossRef]
- Yu, M.; Liu, T.; Chen, Y.; Li, Y.; Li, W. Combination therapy with protein kinase inhibitor H89 and Tetrandrine elicits enhanced synergistic antitumor efficacy. J. Exp. Clin. Cancer Res. 2018, 37, 114. [Google Scholar] [CrossRef]
- Sastry, K.S.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 2017, 36, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Arnal-Estape, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Perron, L.; Bairati, I.; Harel, F.; Meyer, F. Antihypertensive drug use and the risk of prostate cancer (Canada). Cancer Causes Control 2004, 15, 535–541. [Google Scholar] [CrossRef]
- Grytli, H.H.; Fagerland, M.W.; Fossa, S.D.; Tasken, K.A. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Jacobs, E.J.; Deka, A.; Patel, A.V.; Bain, E.B.; Thun, M.J.; Calle, E.E. Use of blood-pressure-lowering medication and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Causes. Control 2009, 20, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.; Carey, I.M.; Owen, C.G.; Harris, T.; Dewilde, S.; Cook, D.G. Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study. Br. J. Clin. Pharmcol. 2011, 72, 157–161. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Rachet, B.; Evans, S.; Smeeth, L. Re: Helene Hartvedt Grytli, Morten Wang Fagerland, Sophie D. Fossa, Kristin Austlid Tasken. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol. In press. http://dx.doi.org/10.1016/j.eururo.2013.01.007. Eur. Urol. 2013, 64, e86–e87. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Medicine (Baltimore) 2015, 94, e1097. [Google Scholar] [CrossRef]
- Emilien, G.; Maloteaux, J.M. Current therapeutic uses and potential of beta-adrenoceptor agonists and antagonists. Eur. J. Clin. Pharmcol. 1998, 53, 389–404. [Google Scholar] [CrossRef]
- Ellison, K.E.; Gandhi, G. Optimising the use of beta-adrenoceptor antagonists in coronary artery disease. Drugs 2005, 65, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Goldstein, D.S. Sympathoneural and adrenomedullary responses to mental stress. Compr. Physiol. 2015, 5, 119–146. [Google Scholar] [PubMed]
- Ullrich, P.M.; Carson, M.R.; Lutgendorf, S.K.; Williams, R.D. Cancer fear and mood disturbance after radical prostatectomy: Consequences of biochemical evidence of recurrence. J. Urol. 2003, 169, 1449–1452. [Google Scholar] [CrossRef]
- Stone, A.A.; Mezzacappa, E.S.; Donatone, B.A.; Gonder, M. Psychosocial stress and social support are associated with prostate-specific antigen levels in men: Results from a community screening program. Health Psychol. 1999, 18, 482–486. [Google Scholar] [CrossRef]
- Turner, E.L.; Lane, J.A.; Metcalfe, C.; Down, L.; Donovan, J.L.; Hamdy, F.; Neal, D.; Vedhara, K. Psychological distress and prostate specific antigen levels in men with and without prostate cancer. Brain Behav. Immun. 2009, 23, 1073–1078. [Google Scholar] [CrossRef]
- Saxe, G.A.; Major, J.M.; Nguyen, J.Y.; Freeman, K.M.; Downs, T.M.; Salem, C.E. Potential attenuation of disease progression in recurrent prostate cancer with plant-based diet and stress reduction. Integr. Cancer 2006, 5, 206–213. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Flores, A.; D’Agostino, R., Jr.; Danhauer, S.C.; Hemal, A.; Kulik, G. A pilot study of blood epinephrine levels and CREB phosphorylation in men undergoing prostate biopsies. Int. Urol. Nephrol. 2014, 46, 505–510. [Google Scholar] [CrossRef]
- White, C.W.; Xie, J.H.; Ventura, S. Age-related changes in the innervation of the prostate gland: Implications for prostate cancer initiation and progression. Organogenesis 2013, 9, 206–215. [Google Scholar] [CrossRef]
- Goepel, M.; Wittmann, A.; Rubben, H.; Michel, M.C. Comparison of adrenoceptor subtype expression in porcine and human bladder and prostate. Urol. Res. 1997, 25, 199–206. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef]
- Cohen, L.; de Moor, C.; Devine, D.; Baum, A.; Amato, R.J. Endocrine levels at the start of treatment are associated with subsequent psychological adjustment in cancer patients with metastatic disease. Psychosom. Med. 2001, 63, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, J.S.; Karavadia, S.S.; Wakefield, M.R. Unusual and underappreciated: Small cell carcinoma of the prostate. Semin. Oncol. 2007, 34, 22–29. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; DeGeest, K.; Sung, C.Y.; Arevalo, J.M.; Penedo, F.; Lucci, J., III; Goodheart, M.; Lubaroff, D.; Farley, D.M.; Sood, A.K.; et al. Depression, social support, and beta-adrenergic transcription control in human ovarian cancer. Brain Behav. Immun. 2009, 23, 176–183. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G.; et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Wingenfeld, K.; Whooley, M.A.; Neylan, T.C.; Otte, C.; Cohen, B.E. Effect of current and lifetime posttraumatic stress disorder on 24-h urinary catecholamines and cortisol: Results from the Mind Your Heart Study. Psychoneuroendocrinology 2015, 52, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Gandubert, C.; Scali, J.; Ancelin, M.L.; Carriere, I.; Dupuy, A.M.; Bagnolini, G.; Ritchie, K.; Sebanne, M.; Martrille, L.; Baccino, E.; et al. Biological and psychological predictors of posttraumatic stress disorder onset and chronicity. A one-year prospective study. Neurobiol. Stress 2016, 3, 61–67. [Google Scholar] [CrossRef]
- Kjeldsen, S.E.; Flaaten, B.; Eide, I.; Helgeland, A.; Leren, P. Evidence of increased peripheral catecholamine release in patients with long-standing, untreated essential hypertension. Scand. J. Clin. Lab. Investig. 1982, 42, 217–223. [Google Scholar] [CrossRef]
- Tsao, P.; von Zastrow, M. Downregulation of G protein-coupled receptors. Curr. Opin. Neurobiol. 2000, 10, 365–369. [Google Scholar] [CrossRef]
- Collins, S.; Caron, M.G.; Lefkowitz, R.J. Regulation of adrenergic receptor responsiveness through modulation of receptor gene expression. Annu. Rev. Physiol. 1991, 53, 497–508. [Google Scholar] [CrossRef]
- Prowatke, I.; Devens, F.; Benner, A.; Grone, E.F.; Mertens, D.; Grone, H.J.; Lichter, P.; Joos, S. Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br. J. Cancer 2007, 96, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Aljameeli, A.; Thakkar, A.; Shah, G. Calcitonin receptor increases invasion of prostate cancer cells by recruiting zonula occludens-1 and promoting PKA-mediated TJ disassembly. Cell. Signal. 2017, 36, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Chigurupati, S.; Anbalagan, M.; Shah, G. Calcitonin increases tumorigenicity of prostate cancer cells: Evidence for the role of protein kinase A and urokinase-type plasminogen receptor. Mol. Endocrinol. 2006, 20, 1894–1911. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, A.B.; Carmena, M.J.; Arenas, M.I.; Bajo, A.M.; Prieto, J.C.; Sanchez-Chapado, M. Overexpression of vasoactive intestinal peptide receptors and cyclooxygenase-2 in human prostate cancer. Analysis of potential prognostic relevance. Histol. Histopathol. 2012, 27, 1093–1101. [Google Scholar] [PubMed]
- Nelson, J.; Bagnato, A.; Battistini, B.; Nisen, P. The endothelin axis: Emerging role in cancer. Nat. Rev. Cancer 2003, 3, 110–116. [Google Scholar] [CrossRef]
- Taub, J.S.; Guo, R.; Leeb-Lundberg, L.M.; Madden, J.F.; Daaka, Y. Bradykinin receptor subtype 1 expression and function in prostate cancer. Cancer Res. 2003, 63, 2037–2041. [Google Scholar] [PubMed]
- Xu, L.L.; Stackhouse, B.G.; Florence, K.; Zhang, W.; Shanmugam, N.; Sesterhenn, I.A.; Zou, Z.; Srikantan, V.; Augustus, M.; Roschke, V.; et al. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res. 2000, 60, 6568–6572. [Google Scholar] [PubMed]
- Sastry, K.S.; Chouchane, A.I.; Wang, E.; Kulik, G.; Marincola, F.M.; Chouchane, L. Cytoprotective effect of neuropeptides on cancer stem cells: Vasoactive intestinal peptide-induced antiapoptotic signaling. Cell Death Dis. 2017, 8, e2844. [Google Scholar] [CrossRef]
- Sastry, K.S.; Smith, A.J.; Karpova, Y.; Datta, S.R.; Kulik, G. Diverse antiapoptotic signaling pathways activated by vasoactive intestinal polypeptide, epidermal growth factor, and phosphatidylinositol 3-kinase in prostate cancer cells converge on BAD. J. Biol. Chem. 2006, 281, 20891–20901. [Google Scholar] [CrossRef]
- Yan, J.; Xiang, J.; Lin, Y.; Ma, J.; Zhang, J.; Zhang, H.; Sun, J.; Danial, N.N.; Liu, J.; Lin, A. Inactivation of BAD by IKK inhibits TNFalpha-induced apoptosis independently of NF-kappaB activation. Cell 2013, 152, 304–315. [Google Scholar] [CrossRef]
- Yancey, D.; Nelson, K.C.; Baiz, D.; Hassan, S.; Flores, A.; Pullikuth, A.; Karpova, Y.; Axanova, L.; Moore, V.; Sui, G.; et al. BAD dephosphorylation and decreased expression of MCL-1 induce rapid apoptosis in prostate cancer cells. PLoS ONE 2013, 8, e74561. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Erb, H.H.; Oh, S.J.; Handle, F.; Feiersinger, G.E.; Luef, B.; Bu, H.; Schafer, G.; Ploner, C.; Egger, M.; et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 2015, 6, 6105–6122. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Tang, J.; Hong, Y.; Song, J.; Terranova, P.F.; Thrasher, J.B.; Svojanovsky, S.; Wang, H.G.; Li, B. Androgen receptor-dependent regulation of Bcl-xL expression: Implication in prostate cancer progression. Prostate 2008, 68, 453–461. [Google Scholar] [CrossRef]
- Krajewska, M.; Krajewski, S.; Epstein, J.I.; Shabaik, A.; Sauvageot, J.; Song, K.; Kitada, S.; Reed, J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar] [PubMed]
- Zellweger, T.; Ninck, C.; Bloch, M.; Mirlacher, M.; Koivisto, P.A.; Helin, H.J.; Mihatsch, M.J.; Gasser, T.C.; Bubendorf, L. Expression patterns of potential therapeutic targets in prostate cancer. Int. J. Cancer 2005, 113, 619–628. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. https://doi.org/10.3390/cancers11030358
Kulik G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers. 2019; 11(3):358. https://doi.org/10.3390/cancers11030358
Chicago/Turabian StyleKulik, George. 2019. "ADRB2-Targeting Therapies for Prostate Cancer" Cancers 11, no. 3: 358. https://doi.org/10.3390/cancers11030358
APA StyleKulik, G. (2019). ADRB2-Targeting Therapies for Prostate Cancer. Cancers, 11(3), 358. https://doi.org/10.3390/cancers11030358