Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Down-Regulating BCL11A in Colorectal Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
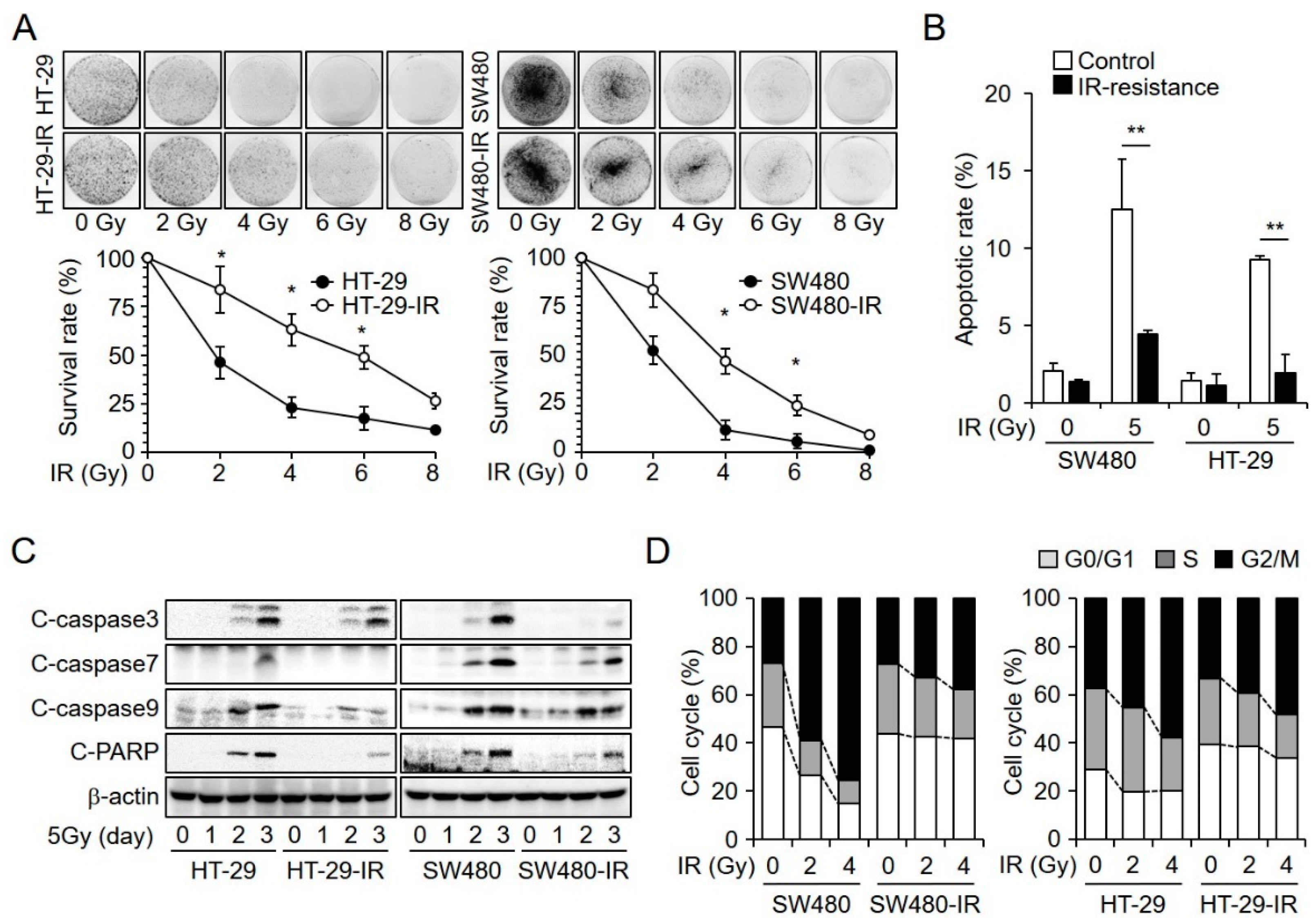
2.1. Establishment and Characterization of Radioresistant CRC Cell Lines
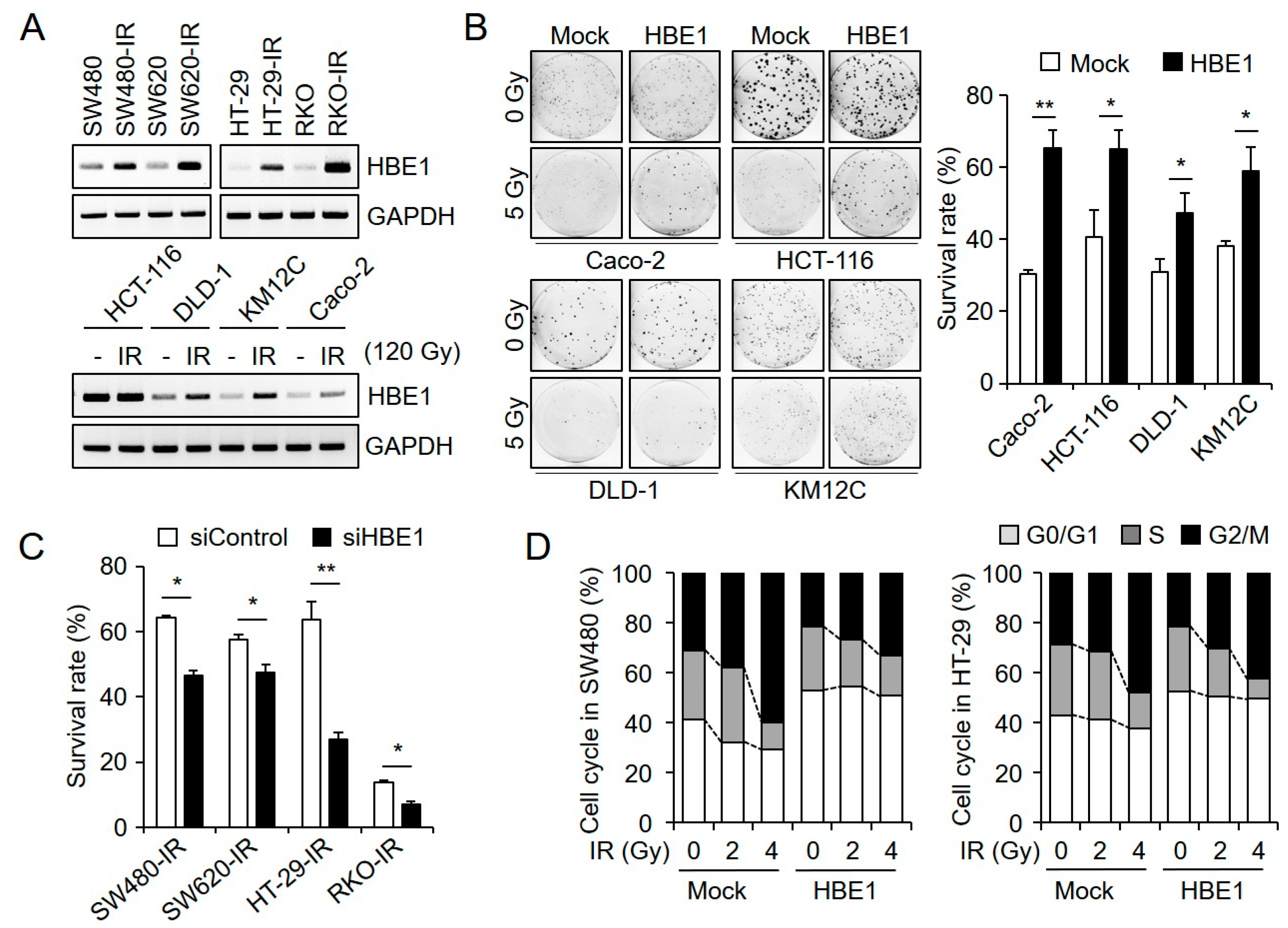
2.2. HBE1 Enhances the Radiation Resistance of CRC Cell Lines
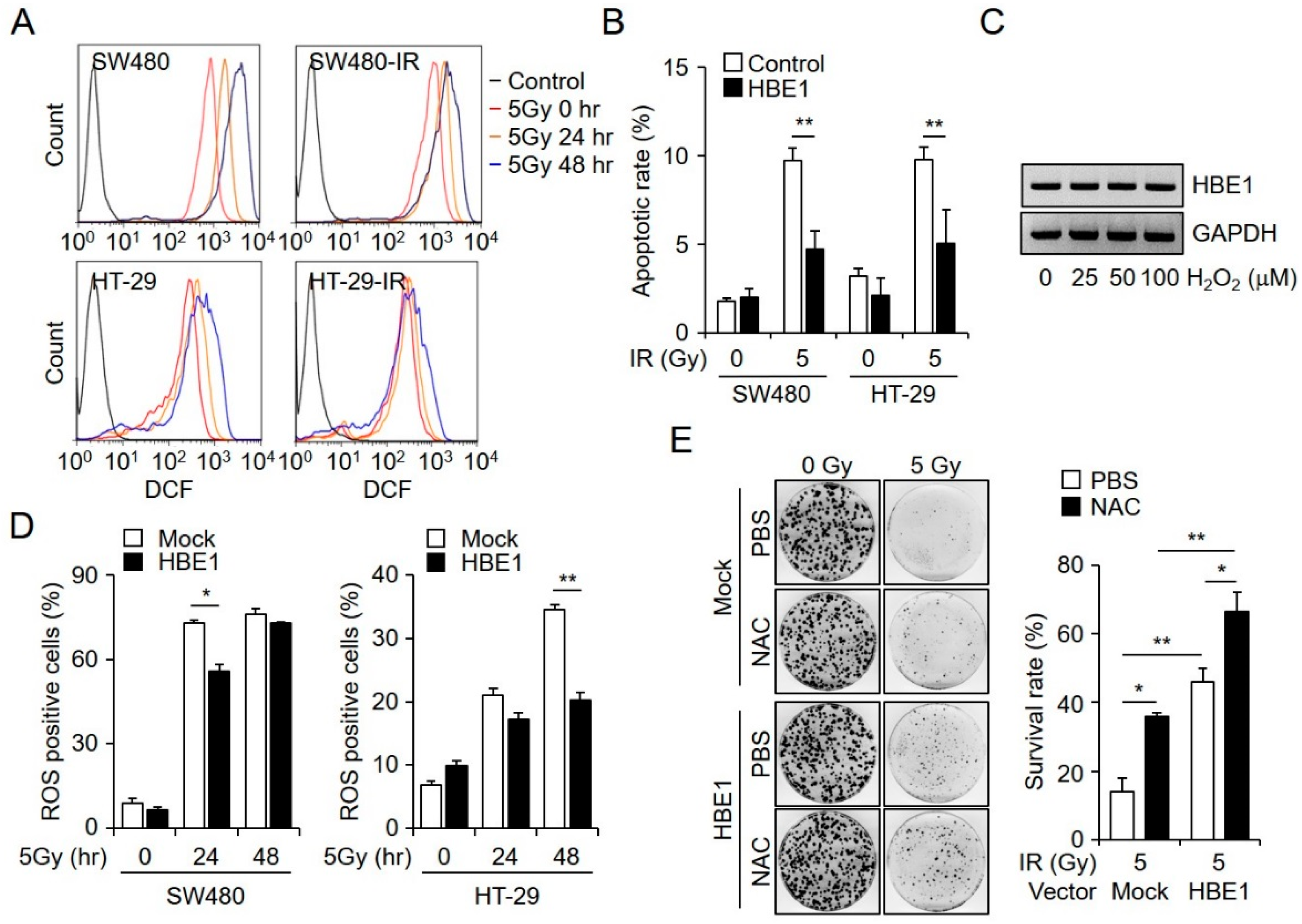
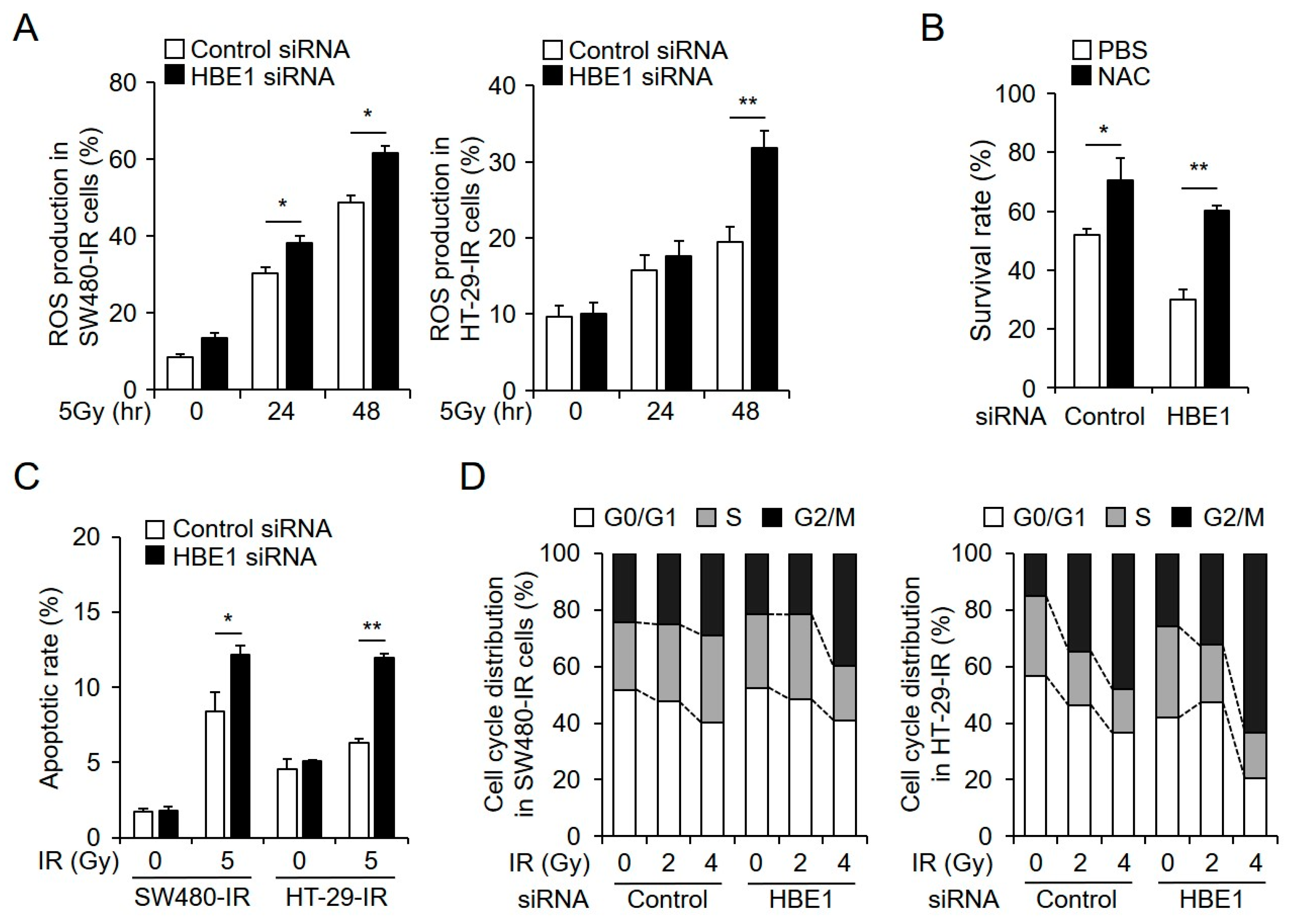
2.3. Radiation-Induced ROS Generation is Regulated by HBE1 Expression
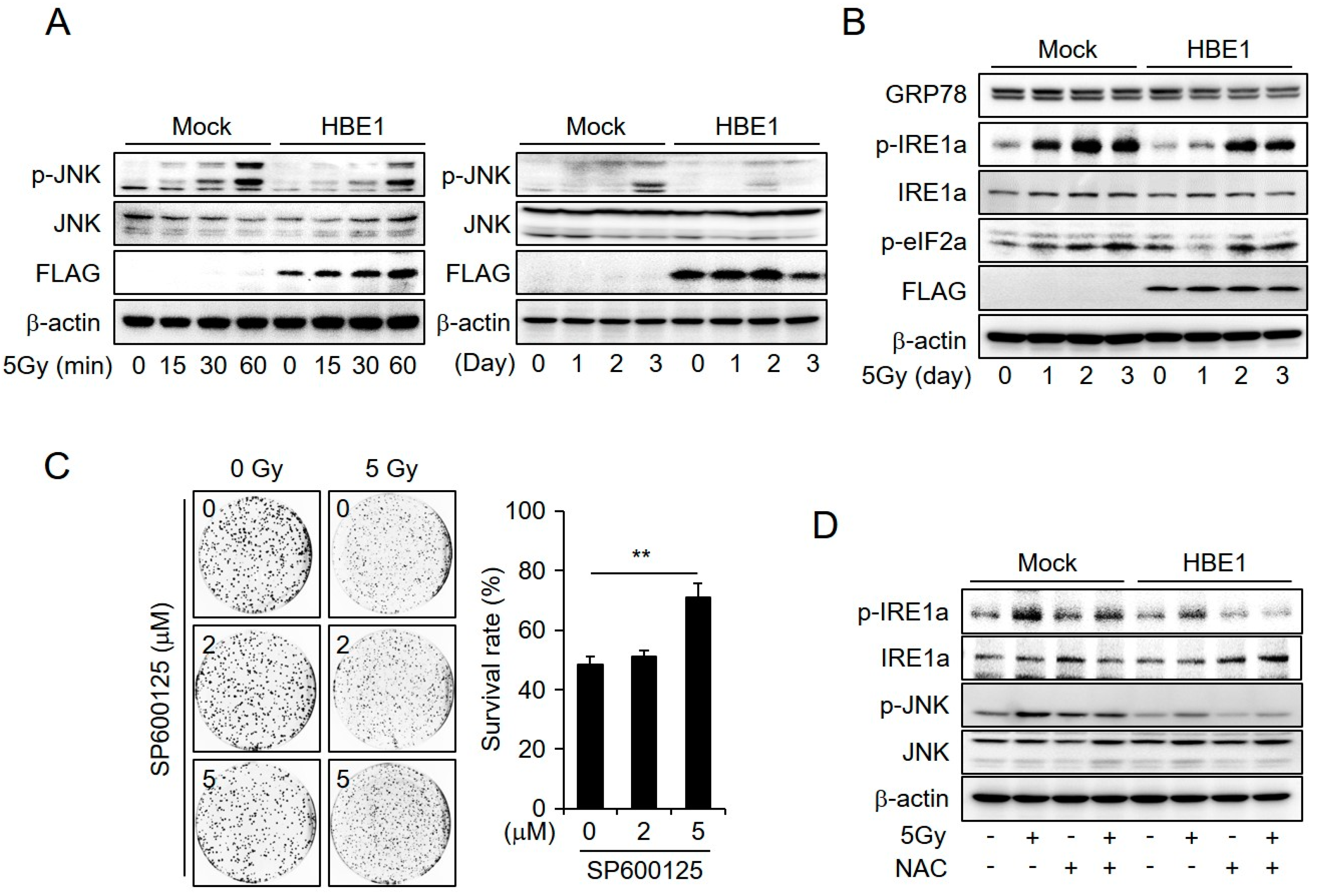
2.4. Enrichment of HBE1 Levels Decreases Radiation-Induced ER-Stress Signaling
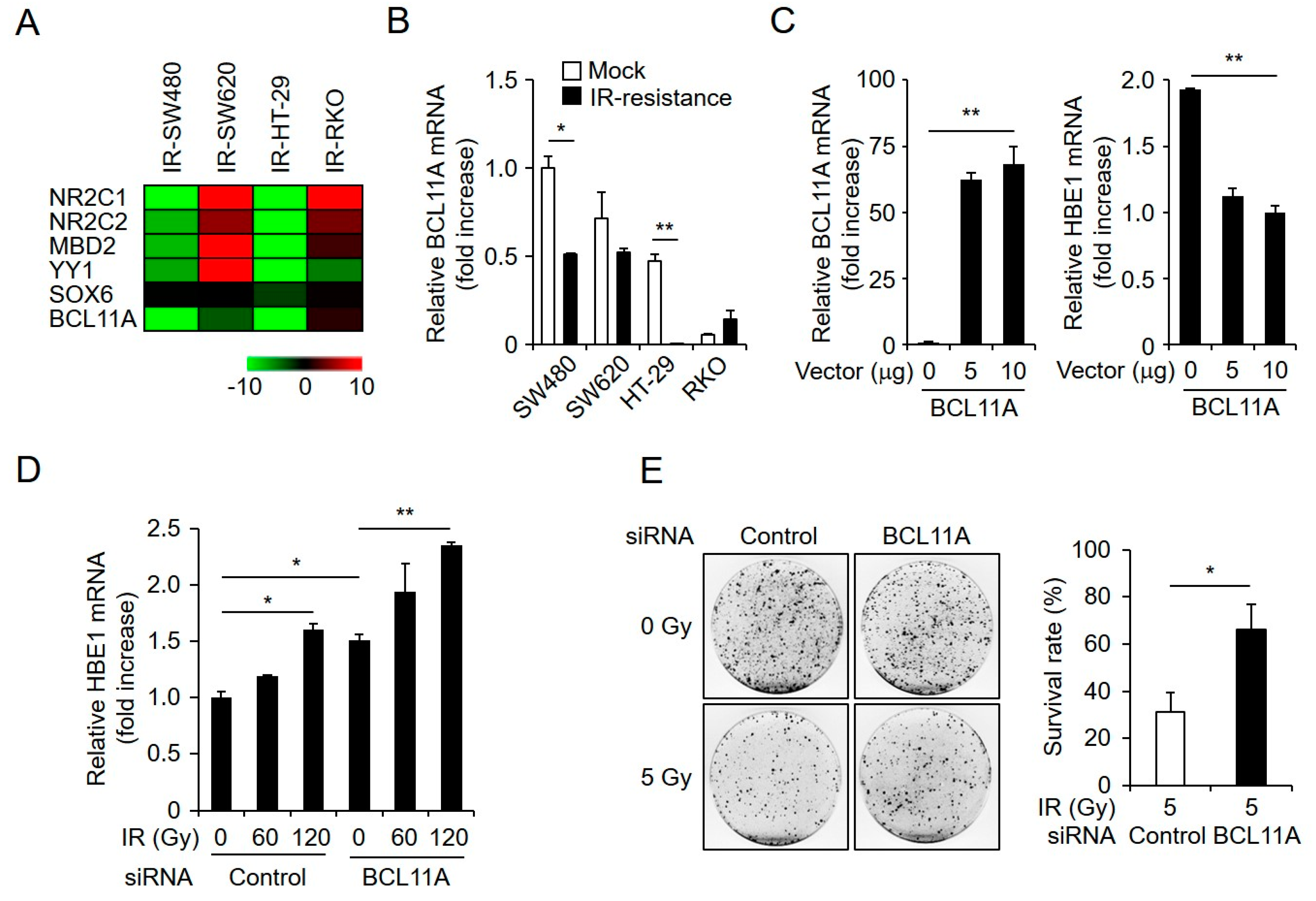
2.5. HBE1 Expression Levels Are Regulated by the Depletion of BCL11A
2.6. HBE1 Deficiency Affects Radiation Sensitivity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Irradiation
4.3. Construction of the HBE1 Expression Plasmid and Transfection
4.4. RNA Interference Experiments
4.5. Western Blot Analysis
4.6. Flow Cytometry for Measurement of ROS Generation and Apoptosis Assay
4.7. Clonogenic Assay
4.8. Cell Cycle Analysis
4.9. Real-Time Quantitative PCR
4.10. Reverse-Transcription PCR
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Habibullah, G.; Gul, R.; Cassum, S.; Elahi, R. Experiences of the Breast Cancer Patients Undergoing Radiotherapy at a Public Hospital Peshawar Pakistan. Asia Pac. J. Oncol. Nurs. 2018, 5, 184–194. [Google Scholar] [CrossRef]
- Folkesson, J.; Birgisson, H.; Pahlman, L.; Cedermark, B.; Glimelius, B.; Gunnarsson, U. Swedish Rectal Cancer Trial: Long lasting benefits from radiotherapy on survival and local recurrence rate. J. Clin. Oncol. 2005, 23, 5644–5650. [Google Scholar] [CrossRef] [PubMed]
- Häfner, M.F.; Debus, J. Radiotherapy for Colorectal Cancer: Current Standards and Future Perspectives. Visc. Med. 2016, 32, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.B.; Sheng, X.; Zhang, N.; Yang, M.W.; Wang, F. Role of microRNAs in the resistance of colorectal cancer to chemoradiotherapy. Mol. Clin. Oncol. 2018, 8, 528–532. [Google Scholar] [CrossRef]
- Hur, W.; Yoon, S.K. Molecular Pathogenesis of Radiation-Induced Cell Toxicity in Stem Cells. Int. J. Mol. Sci. 2017, 18, 2749. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. (R. Coll. Radiol.) 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, J.N.; Redmond, K.M.; Schettino, G.; Prise, K.M. DNA double strand break repair: A radiation perspective. Antioxid. Redox Signal. 2013, 18, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Advani, S.J.; Lawson, J.D.; Kahle, K.T.; Ng, K.; Carter, B.; Chen, C.C. DNA damage response and repair: Insights into strategies for radiation sensitization of gliomas. Future Oncol. 2011, 7, 1335–1346. [Google Scholar] [CrossRef]
- Borges, H.L.; Linden, R.; Wang, J.Y. DNA damage-induced cell death: Lessons from the central nervous system. Cell Res. 2008, 18, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef]
- Maryam, K.A.; Dariush, M.; Azadeh, S.Z.; Maryam, E.G.; Abbas, M.; Asefeh, H.; Sohrab, H.; Ali, B.; Hadi, P. Pre and post radiotherapy serum oxidant/antioxidant status in breast cancer patients: Impact of age, BMI and clinical stage of the disease. Rep. Pract. Oncol. Radiother. 2016, 21, 141–148. [Google Scholar] [CrossRef]
- Woźniak, A.; Masiak, R.; Szpinda, M.; Mila-Kierzenkowska, C.; Woźniak, B.; Makarewicz, R.; Szpinda, A. Oxidative stress markers in prostate cancer patients after HDR brachytherapy combined with external beam radiation. Oxid. Med. Cell. Longev. 2012, 2012, 789870. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene 2010, 29, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef] [PubMed]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Çimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Kamimura, D.; Bevan, M.J. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. 2008, 181, 5433–5441. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [PubMed]
- Miccio, A.; Blobel, G.A. Role of the GATA-1/FOG-1/NuRD pathway in the expression of human beta-like globin genes. Mol. Cell Biol. 2010, 30, 3460–3470. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, Z.; Russell, J.E. A human embryonic hemoglobin inhibits Hb S polymerization in vitro and restores a normal phenotype to mouse models of sickle cell disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10635–10640. [Google Scholar] [CrossRef]
- Wang, Y.; Rank, G.; Li, Z.; Wang, Y.; Ju, J.; Nuber, A.; Wu, Y.; Liu, M.; Nie, M.; Huang, F.; et al. ε-globin expression is regulated by SUV4-20h1. Haematologica 2016, 101, e168–e172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Nakarai, C.; Osawa, K.; Akiyama, M.; Matsubara, N.; Ikeuchi, H.; Yamano, T.; Hirota, S.; Tomita, N.; Usami, M.; Kido, Y. Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clin. Exp. Med. 2015, 15, 333–341. [Google Scholar] [CrossRef]
- Rahmanian, N.; Hosseinimehr, S.J.; Khalaj, A. The paradox role of caspase cascade in ionizing radiation therapy. J. Biomed. Sci. 2016, 23, 88. [Google Scholar] [CrossRef]
- Fernet, M.; Mégnin-Chanet, F.; Hall, J.; Favaudon, V. Control of the G2/M checkpoints after exposure to low doses of ionising radiation: Implications for hyper-radiosensitivity. DNA Repair (Amst.) 2010, 9, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Jo, G.H.; Bögler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef]
- Hellweg, C.E.; Spitta, L.F.; Henschenmacher, B.; Diegeler, S.; Baumstark-Khan, C. Transcription Factors in the Cellular Response to Charged Particle Exposure. Front. Oncol. 2016, 6, 61. [Google Scholar] [CrossRef]
- Tanabe, O.; Katsuoka, F.; Campbell, A.D.; Song, W.; Yamamoto, M.; Tanimoto, K.; Engel, J.D. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 2002, 21, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Raich, N.; Clegg, C.H.; Grofti, J.; Roméo, P.H.; Stamatoyannopoulos, G. GATA1 and YY1 are developmental repressors of the human epsilon-globin gene. EMBO J. 1995, 14, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Cohen-Barak, O.; Hagiwara, N.; Kingsley, P.D.; Fuchs, D.A.; Erickson, D.T.; Epner, E.M.; Palis, J.; Brilliant, M.H. Sox6 directly silences epsilon globin expression in definitive erythropoiesis. PLoS Genet. 2006, 2, e14. [Google Scholar] [CrossRef] [PubMed]
- Rupon, J.W.; Wang, S.Z.; Gnanapragasam, M.; Labropoulos, S.; Ginder, G.D. MBD2 contributes to developmental silencing of the human ε-globin gene. Blood Cells Mol. Dis. 2011, 46, 212–219. [Google Scholar] [CrossRef]
- Chan, K.S.; Xu, J.; Wardan, H.; McColl, B.; Orkin, S.; Vadolas, J. Generation of a genomic reporter assay system for analysis of γ- and β-globin gene regulation. FASEB J. 2012, 26, 1736–1744. [Google Scholar] [CrossRef]
- Grundmann, O.; Mitchell, G.C.; Limesand, K.H. Sensitivity of salivary glands to radiation: From animal models to therapies. J. Dent. Res. 2009, 88, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.G.; Stone, H.B.; Wong, R.S.; Capala, J.; Bernhard, E.J.; Vikram, B.; Coleman, C.N. Normal tissue protection for improving radiotherapy: Where are the Gaps? Transl. Cancer Res. 2012, 1, 35–48. [Google Scholar]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegué, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Willers, H.; Dahm-Daphi, J.; Powell, S.N. Repair of radiation damage to DNA. Br. J. Cancer 2004, 90, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Chui, D.H.; Fucharoen, S.; Chan, V. Hemoglobin H disease: Not necessarily a benign disorder. Blood 2003, 101, 791–800. [Google Scholar] [CrossRef]
- Saha, D.; Patgaonkar, M.; Shroff, A.; Ayyar, K.; Bashir, T.; Reddy, K.V. Hemoglobin expression in nonerythroid cells: Novel or ubiquitous? Int. J. Inflam. 2014, 803237. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Akaishi, J.; Asaka, S.; Okamoto, J.; Miyamoto, S.; Mizutani, K.; Yoshida, A.; Ito, K.; Emi, M. Decreased expression of haemoglobin beta (HBB) gene in anaplastic thyroid cancer and recovery of its expression inhibits cell growth. Br. J. Cancer 2005, 92, 2216–2224. [Google Scholar] [CrossRef]
- Nishi, H.; Inagi, R.; Kato, H.; Tanemoto, M.; Kojima, I.; Son, D.; Fujita, T.; Nangaku, M. Hemoglobin is expressed by mesangial cells and reduces oxidant stress. J. Am. Soc. Nephrol. 2008, 19, 1500–1508. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Liu, L.; Wen, X.; Xu, Y.; Chen, J.; Teng, J. Cab45S inhibits the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis. 2014, 5, e1219. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef]
- Shimura, T.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016, 15, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Carmia, B. Antioxidants and Radiation Therapy. J. Nutr. 2004, 134, 3207S–3209S. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef]
- Lamson, D.W.; Brignall, M.S. Antioxidants in cancer therapy; their actions and interactions with oncologic therapies. Altern. Med. Rev. 1999, 4, 304–329. [Google Scholar] [PubMed]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with β-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 14, 313–326. [Google Scholar] [CrossRef]
- Liu, P.; Keller, J.R.; Ortiz, M.; Tessarollo, L.; Rachel, R.A.; Nakamura, T.; Jenkins, N.A.; Copeland, N.G. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003, 4, 525–532. [Google Scholar] [CrossRef]
- Kamimura, K.; Mishima, Y.; Obata, M.; Endo, T.; Aoyagi, Y.; Kominami, R. Lack of Bcl11b tumor suppressor results in vulnerability to DNA replication stress and damages. Oncogene 2007, 26, 5840–5850. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, H.; He, D.; Hu, X.; Li, Y. Downregulation of BCL11A by siRNA induces apoptosis in B lymphoma cell lines. Biomed. Rep. 2013, 1, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, E.; Sonoki, T.; Willis, T.G.; Harder, L.; Nowak, R.; Arriola, E.L.; Liu, H.; Price, H.P.; Gesk, S.; Steinemann, D.; et al. The BCL11 gene family: Involvement of BCL11A in lymphoid malignancies. Blood 2001, 98, 3413–3420. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Lee, S.-J.; Cho, H.J.; Kim, J.-T.; Yoon, H.R.; Lee, K.H.; Kim, B.Y.; Lee, Y.; Lee, H.G. Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Down-Regulating BCL11A in Colorectal Cancer Cells. Cancers 2019, 11, 498. https://doi.org/10.3390/cancers11040498
Park SY, Lee S-J, Cho HJ, Kim J-T, Yoon HR, Lee KH, Kim BY, Lee Y, Lee HG. Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Down-Regulating BCL11A in Colorectal Cancer Cells. Cancers. 2019; 11(4):498. https://doi.org/10.3390/cancers11040498
Chicago/Turabian StylePark, Sang Yoon, Seon-Jin Lee, Hee Jun Cho, Jong-Tae Kim, Hyang Ran Yoon, Kyung Ho Lee, Bo Yeon Kim, Younghee Lee, and Hee Gu Lee. 2019. "Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Down-Regulating BCL11A in Colorectal Cancer Cells" Cancers 11, no. 4: 498. https://doi.org/10.3390/cancers11040498
APA StylePark, S. Y., Lee, S.-J., Cho, H. J., Kim, J.-T., Yoon, H. R., Lee, K. H., Kim, B. Y., Lee, Y., & Lee, H. G. (2019). Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Down-Regulating BCL11A in Colorectal Cancer Cells. Cancers, 11(4), 498. https://doi.org/10.3390/cancers11040498