Abstract
Cancers cells have the ability to develop chemotherapy resistance, which is a persistent problem during cancer treatment. Chemotherapy resistance develops through different molecular mechanisms, which lead to modification of the cancer cells signals needed for cellular proliferation or for stimulating an immune response. The endoplasmic reticulum (ER) is an important organelle involved in protein quality control, by promoting the correct folding of protein and ER-mediated degradation of unfolded or misfolded protein, namely, ER-associated degradation. Disturbances of the normal ER functions causes an accumulation of unfolded or misfolded proteins in the ER lumen, resulting in a condition called “ER stress (ERS).” ERS triggers the unfolded protein response (UPR)—also called the ERS response (ERSR)—to restore homeostasis or activate cell death. Although the ERSR is one emerging potential target for chemotherapeutics to treat cancer, it is also critical for chemotherapeutics resistance, as well. However, the detailed molecular mechanism of the relationship between the ERSR and tumor survival or drug resistance remains to be fully understood. In this review, we aim to describe the most vital molecular mechanism of the relationship between the ERSR and chemotherapy resistance. Moreover, the review also discusses the molecular mechanism of ER stress-mediated apoptosis on cancer treatments.
1. Introduction
Cancer is one of the leading causes of death, worldwide, for instance there were about 15 million diagnosed cancer cases and 8.2 million deaths in 2013 [1]. Cancer is the second leading cause of death for amongst the United States population, after heart disease, and the leading cause of death for non-Hispanic, Asian, or Pacific Islander, and Hispanic populations [2]. According to the Korea National Statistical Office, cancer (malignant neoplasms) was significantly higher than deaths caused by cerebrovascular disease, heart disease, diabetes, suicide, and other deaths [3]. Like getting rid of weeds, healing from cancer becomes more and more difficult, day by day. The treatments that kill cancer cells are generally toxic to normal cells, as well [4]. The main objective of cancer treatment is to destroy cancer cells, while causing minimal damage to normal tissue, which can be achieved, either directly or indirectly, by modifying the signals needed for cellular proliferation in cancer cells or by stimulating an immune response [5,6]. The therapeutic management of cancer depends on the cancer type, its location and extent, the patient age, and other characteristics, including specific pathological, molecular, genetic, epigenetic, and microenvironmental changes in which the cancer cell resides [7,8,9,10]. Cancers can be treated with a combination of therapies (surgery, radiation, chemotherapy, laser therapy, and targeted therapy), chosen on the basis of the type and stage of cancer [11]. Cancer cells have the ability to develop resistance to chemotherapeutics, which is a persistent problem during cancer treatment [6,12]. Chemotherapy becomes resistant through different mechanisms, including patho-physiological, micro-environmental, genetic, and epigenetic changes in the tumor cell [13]. The increasing prevalence of chemotherapy resistance requires the development of further treatments and effective research.
The endoplasmic reticulum (ER) has multifunctional activities, including protein folding, protein maturation, and ER quality control (ERQC), to maintain a cellular homeostasis [14]. The perturbation of the normal ERQC system causes an accumulation of unfolded or misfolded proteins in the ER lumen, resulting in a condition called “ER stress (ERS)” [15]. Upon ERS, endoplasmic reticulum stress response (ERSR) is produced to restore homeostasis or activate cell death [16]. Several studies have suggested that the ERSR could be the potential target for chemotherapeutics to treat cancer [17,18]. Recently, it has been reported that the ERS is critical for chemo-therapeutics resistance, following the initiation of an ERSR [19,20,21,22]. Although a thorough understanding of the ERSR associated with cancer drug resistance will enable us to develop more effective chemotherapeutic candidates, the relationship between the ERSR and chemotherapy resistance remains poorly understood. In this review, we focused on the detailed molecular mechanism of the relationship between ER stress and tumor survival or drug resistance. In addition, the effects of ER stress-mediated apoptosis on cancer treatments are also presented.
2. Apoptosis as a Therapeutic Target for Anticancer Therapy
Chemotherapy, radiation therapy, and surgery are the main procedures associated with cancer management. The main objective of cancer therapy is to destroy all the cancer cells, while causing minimal damage to the normal tissue. Apoptosis or the process of programmed cell death is a genetically regulated form of cell death, and is an emerging target for anticancer therapy [23]. In recent years, the most vital advances in clinical oncology involve the killing of tumor cells, mostly by apoptosis, which crucially determines the treatment response. For example, current cancer therapies including chemotherapy, radiotherapy, suicide gene therapy, or immunotherapy, exhibit antitumor effects by activating the apoptosis signal transduction pathways in cancer cells [24]. There are three different pathways that lead to apoptosis, which are (a) the extrinsic death receptor pathway, (b) the intrinsic mitochondrial pathway, and (c) the ERS pathway (Figure 1). These pathways are activated by both intracellular and extracellular signals and converge at the executioner caspases, to carry out apoptosis through the cleavage of hundreds of proteins [25].
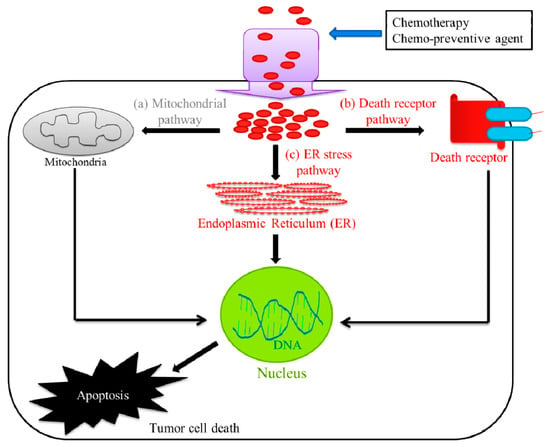
Figure 1.
Pathways involved in tumor cell death. The chemotherapeutic treatment or chemotherapy mainly utilize three different pathways that lead to tumor cell death or apoptosis, which are (a) the extrinsic death receptor pathway, (b) the intrinsic mitochondrial pathway, and (c) the endoplasmic reticulum stress (ERS) pathway.
2.1. Extrinsic Death Receptor Pathway
The extrinsic pathway (death-receptor-dependent), belongs to the superfamily of tumor necrosis factor receptors (TNFR), while their respective protein TNF family ligands activate caspases, via cell-surface death receptors, which respond to the cognate death ligands expressed on immune-effector cells [26,27]. The extrinsic apoptosis pathway is an attractive target to trigger apoptosis in tumor cells for cancer therapy, as death receptors have been directly linked with the cell’s death machinery [28].
2.2. Intrinsic Mitochondrial Pathways
The intrinsic or death receptor apoptotic pathway activates caspases via members of the B-cell lymphoma-2 (Bcl-2) protein family and the mitochondria, as a reaction to severe cellular damage or stress [26]. Mitochondria play a vital role in cellular processes, such as cellular energy metabolism, cell differentiation, and regulation of the cell cycle. In addition, mitochondria control apoptosis through the regulated suppression or activation of pro-apoptotic proteins, electron transport control, and maintenance of cellular redox potential [29,30]. Mitochondria are targets with great potential as therapeutics for cancer. Mitochondrial dysfunction activates apoptotic processes that lead to changes in the mitochondrial structure, loss of electron transport function, and disruption of the mitochondrial membrane potential, leading to the release of reactive oxygen species (ROS), caspase activation, and activation of other primary mediators of apoptosis [31,32].
2.3. Endoplasmic Reticulum Stress Pathways
ER is an important organelle involved in multiple cellular activities, including synthesis, maturation, translation, and folding of secretory and membrane proteins, lipid biogenesis, and the sequestration of Ca2+ [33,34,35]. Various physiological and pathological conditions might affect ER homeostasis, causing ERS. The homeostatic mechanisms that maintain the balance of large numbers of proteins are ultimate targets of novel anticancer strategies [36]. Actually, ER signaling pathways are an emerging target for cancer therapeutics. ERS is of great interest to cancer biologists, as both the intrinsic mitochondrial and ER stress-mediated apoptosis pathways are activated by each other’s mechanisms.
3. Endoplasmic Reticulum Stress-Mediated Apoptosis and Cancer Therapy
ERS is used as an emerging therapeutic target for cancer treatment. Recently, a number of major hallmarks of cancer have been identified that are expected to facilitate the development of anticancer therapies. For example, a drug combination of rapamycin and the heat shock protein 90 (HSP90) inhibitor retaspimycin, optimizes ERS and proteotoxicity, leading to the suppression of uncontrollable rat sarcoma (RAS)-driven tumors [37].
Cellular adaptation to ERS is mediated by the UPR or ERSR, which works to restore the ER homeostasis. The cancer therapeutics targeted ERS-mediated apoptosis, through the ERSR, to kill tumor cells. For example, Osajin, a prenylated isoflavone, activates ERSR and the ER-resident cysteine protease, caspase-12, leading to caspase-3 activation and apoptosis in human nasopharyngeal carcinoma (NPC) [38]. The ERSR is mainly transduced by three ER-resident sensor proteins, (i) protein kinase RNA-like endoplasmic reticulum kinase (PERK), (ii) activating transcription factor 6 alpha (ATF6), and (iii) inositol-requiring enzyme 1 alpha (IRE1α) [39,40]. The luminal domains of these sensor proteins bind an ER chaperone, binding-immunoglobulin protein (BiP), that are separated at ERS conditions, which lead to the activation of ERSR [33]. The integrated signaling downstream of these three sensors, tightly controls life-or-death decisions in the cells exposed to ERS (Figure 2). The ERSR can induce pro-apoptotic signaling through the activation of DNA damage-inducible transcript 3, also known as the C/EBP homologous protein (CHOP), which subsequently leads to the initiation of apoptosis [41]. The CHOP can induce ERS-mediated apoptosis, through the down-regulation of anti-apoptotic Bcl-2 protein, and the upregulation of Bcl-2-associated X (Bax) protein [42,43]. Bcl-2 and Bax control a diversity of cellular responses to ERS, through the transport of Ca2+, in and out of the ER lumen. The Ca2+ released from ER through inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) at ER and mitochondrial contact sites, can promote apoptosis, mainly via the release of cytochrome c from the mitochondria [44,45]. The cytochrome c then interacts with the apoptosis protease-activating factor-1 (Apaf-1), adenosine triphosphate (ATP), and pro-caspase-9 to form a supramolecular complex called the apoptosome, subsequently activating the caspase pathway through autocatalysis, and ultimately activating caspase-3, resulting in apoptosis (Figure 2) [46,47].
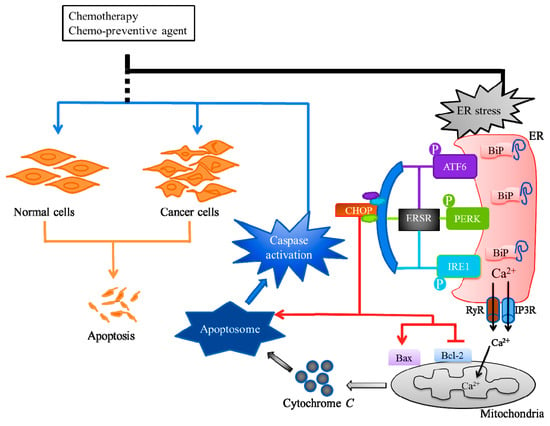
Figure 2.
Endoplasmic reticulum stress (ERS)-mediated apoptosis and cancer therapy. Cancers can be treated with radiotherapy or chemotherapy, either individually or as a combination therapy. The chemotherapeutic exerts its anticancer activity by inducing ERS through endoplasmic reticulum stress response (ERSR) to kill tumor cells. The ERSR is mainly transduced by three ER-resident sensor proteins, protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating the transcription factor 6 alpha (ATF6), and inositol requiring enzyme 1 alpha (IRE1). The integrated signaling downstream of these three sensors can induce pro-apoptotic signaling through the activation of C/EBP homologous protein (CHOP) that can downregulate B-cell lymphoma-2 (Bcl-2) protein and upregulate Bcl-2-associated X (Bax) protein. The Ca2+ released through inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) at ER and mitochondrial contact sites can promote the release of mitochondria cytochrome c which interacts with apoptosis protease-activating factor-1 (Apaf-1), adenosine triphosphate (ATP) and procaspase-9 to form the apoptosome, and subsequently activates the caspase pathway resulting in apoptosis.
4. Chemotherapy Resistance: Role of Endoplasmic Reticulum Stress Response (ERSR)
4.1. Chemotherapy Resistance
Drug resistance refers to the reduction of the effectiveness of drugs, including antibiotics, antimalarial, antiviral, and chemotherapeutic agents, through different mechanisms during the treatment of various diseases [48]. Chemotherapy resistance is the tolerance of tumor cells to cancer treatments, such as chemotherapy, and targeted therapies. Chemotherapy resistance can be primary or acquired, which result in lowering drug accumulation and increasing drug efflux, or alterations of drug targets and signaling transduction molecules, leading to the repair of drug-induced DNA damage, subsequently blocking apoptosis, to kill tumor cells [49]. Primary or innate drug resistance occurs prior to any given treatment, whereas acquired resistance develops after the initial therapy [49]. Therapeutic resistance is a major barrier to the improvement of outcomes, for a variety of cancers [50].
The underlying mechanisms of cancer drug resistance are complex, and involve alteration in drug transports and targets, modification of signaling transduction molecules, or reactivation of antioxidant systems, leading to an impaired apoptosis.
Altered Membrane Transport
Drug transport across biological membranes is possible through passive trans-cellular transport and carrier-mediated transporters. Carrier-mediated transporters are of great interest to cancer researchers, as the transporters are responsible for the uptake, accumulation, distribution, and the efflux of drugs [51]. As examples, drug influx transporters including organic anion transporter (OAT), organic anion-transporting polypeptide (OATP), organic cation transporter (OCT), novel organic cation-transporter (OCTN), oligopeptide transporter (PEPT), and copper transporter (Ctr), are responsible for the uptake of most antineoplastic drugs [52,53,54]. The ATP-dependent multidrug transporters are in the class of carrier-mediated transporters, which are in the ubiquitous superfamily of ATP-binding cassette (ABC) proteins. ABC proteins, like multidrug resistance protein (MRP) and breast cancer resistance protein (BCRP) actively efflux chemotherapeutic agents out of the cell [55,56,57,58,59].
Alteration of the membrane transport system is one of the most important factors of cancer therapeutic resistance, in which the membrane proteins extrude chemotherapeutic agents, leading to a lower concentration of the drugs required to kill the target cells, and ultimately, a failure of chemotherapy [60]. As an example, the over-expression of ABC transporters on the plasma membrane can promote drug resistance in cancer cells [61]. ABC transporters have also been linked to resistance to infectious diseases, such as acquired immune deficiency syndrome (AIDS) and malaria [62,63]. MRP1 has been upregulated in different tumors and overexpressed in various cancer cells, during anticancer treatments [64,65]. MRP1 transporters were found to be up-regulated in colon cancer, which can facilitate the efflux of anticancer drugs out of cancer cells and decrease their therapeutic effects [66,67]. In addition to their physiological roles in host detoxification and pharmacokinetics, dysregulation of ABC transporters is associated with a variety of diseases.
4.2. Alteration of Drug Targets
Alteration of the molecular target significantly influences the effectiveness of cancer therapy, such as mutations or modifications of gene expression levels, leading to drug resistance. For example, active anticancer drugs, notably etoposide and doxorubicin-targeting DNA topoisomerase II (Topo II), play critical roles in DNA replication, transcription, and chromosome segregation [68,69,70,71]. Topoisomerase-targeting anticancer drugs act through (i) topoisomerase poisoning, which leads to a replication fork arrest and double-strand break formation, and (ii) topoisomerase inhibition, which leads to inhibition of the ATPase catalytic activity. The cancer cells can resist cancer therapy through various means. For example, some cancer cells have become resistant to Topo II-inhibiting drugs, through mutations in the Topo II gene, and some by means of increasing or decreasing its activities [72,73,74]. MRP1 confers resistance to Topo II-poisoning drugs (e.g., doxorubicin and etoposide), which is associated with a decreased cellular accumulation of cytotoxic drugs [75,76].
4.3. Activation of Antioxidant and Detoxification Systems
Chemotherapy-resistant cancer cells show a high expression of antioxidants and detoxifying systems. Most anticancer drugs require metabolic activation to achieve clinical efficacy. This means that the cancer cells can develop resistance through decreasing drug activation. For example, the anticancer drugs doxorubicin and oracin are inactivated by aldo-keto reductase (AKR) 1C3 [77]. Similar to oracin, doxorubicin goes through a metabolic detoxification by carbonyl reduction to the corresponding C13 alcohol metabolite [78]. The platinum-based cancer drug encounters resistance through the activation of detoxification systems by metallothionein and thiol glutathione [79]. Glutathione (GSH) and GSH-metabolism play an important role in cancer prevention and progression. GSH depletion has been shown to sensitize cells to compounds that produce oxidative cytolysis [80]. Elevation of Glutathione S-transferases (GST), which are also associated with resistance to apoptosis, can be stimulated by a variety of stimuli [81]. GSTs also play a role in the development of drug resistance through direct detoxification, by inhibition of the mitogen-activated protein kinase (MAPK) pathway [82].
4.4. Endoplasmic Reticulum Stress Response (ERSR) Is one of the Major Signaling Pathways of Chemotherapy Resistance
ER is an essential site of cellular homeostasis regulation, which can be hampered by various physiological and pathological conditions, resulting in ERS [83]. Recently, it has been demonstrated that cancer cells with constitutive or acquired resistance to chemotherapy are also resistant to ERS-triggered cell death [19]. The ERSR is a pathway of recent interest to many investigators, due to its role in adaptive survival signaling in cancer. The ERSR represents an adaptive mechanism that supports survival and chemo-resistance of tumor cells. The ERSR is regulated by ATF6, IRE1, and PERK in normal cells [84], while it is often deregulated and promotes tumorigenicity, as the depletion of tumor suppressors or activation of oncogenes, favors cells that survive during high protein synthesis and metabolic stress in cancer cells [85].
4.4.1. Role of Protein Kinase RNA-Like ER Kinase (PERK) in Chemotherapy Resistance
ERSR or UPR is initiated in cancer cells, through three ER sensors ATF6, IRE1, and PERK [86,87]. ERS-mediated PERK-dependent signaling is critical for cell survival or drug resistance, following the initiation of an ERSR (Figure 3). For example, the survival rate has been dramatically reduced in PERK-deleted embryonic stem cells, following exposure to ERS-inducing agents [88]. The ERS-mediated activation PERK phosphorylates eukaryotic translation initiation factor-2α (eIF2α) and nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2), leading to the attenuation of protein translation and an increase of genes controlling the redox homeostasis [87,89].
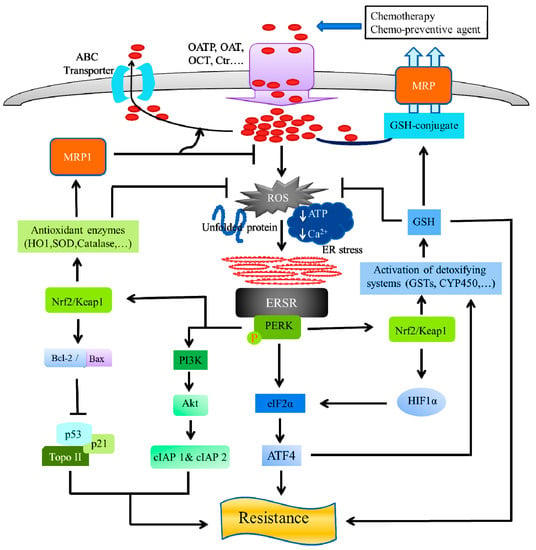
Figure 3.
ER stress and chemo-resistance: Role of protein kinase R-like endoplasmic reticulum kinase (PERK). The anti-neoplastic drugs, possess ERS-mediated apoptosis and exert their anticancer effect by entering the tumor cells, using influx transporters, including an organic anion transporter (OAT), organic anion-transporting polypeptide (OATP), organic cation transporter (OCT), and a copper transporter (Ctr). On the other hand, ATP-dependent multidrug transporters of the carrier-mediated, ubiquitous superfamily of ATP-binding cassette (ABC) proteins actively efflux chemotherapeutic agents out of the cell. ABC proteins include the multidrug resistance protein (MDR), which confer resistance to many anticancer drugs in cancer cells. The ERS-mediated activated PERK, phosphorylates eukaryotic translation initiation factor-2α (eIF2α) and nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2). Activation of PERK via agents that trigger the ERSR, promotes a dissociation of the cytoplasmic Nrf2/Keap1, and the Nrf2 possess its cyto-protective activity by activating detoxifying enzyme systems, including the cytochrome P450 (CYP) system, glutathione-S-transferases (GSTs), thus, reducing ROS, leading to the prevention of apoptosis in cancer cells. The GSTs ensure a high GSH concentration and GSH-transporters inside cancer cells, which are associated with a high resistance to chemotherapeutic agents. The GSH exhibits cancer cell resistance to anti-neoplastic treatments, either by inhibition of apoptosis or chemotherapeutic drug detoxification, by the GSH-drug conjugation, followed by efflux through the GSH-pump of MRP transporters. The activated PERK-Nrf2 signaling, upregulates the multidrug resistance protein 1 (MRP1), by the activation of antioxidant enzymes (HO-1, SOD, catalase, etc.) that neutralize the reactive oxygen species (ROS) and increases the drug efflux, leading to a reduction in the ROS levels. Nrf2 stabilization by the exposure of antioxidant enzymes, leads to an increased Bcl-2/Bax ratio, subsequently reducing apoptosis and increasing cell survival, by regulation of Topo II, p53, and its target gene p21. The PERK activity can induce cellular inhibitor of apoptosis (cIAP1 and cIAP2) proteins, by the activation of phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway, leading to chemotherapy resistance.
Most of the biological activities of PERK have been initiated through its downstream target translational initiation factor eIF2α kinase and subsequent translational upregulation of ATF4. A cell with compromised PERK/eIF2α/ATF4 signaling confers a survival advantage of tumor cells, under hypoxia [90]. Hypoxia is a unique feature of the tumor cells that contributes to cancer’s progression. To overcome hypoxic stress, tumor cells initiate hypoxia-inducing factor (HIF)-dependent transcriptional responses, through hypoxia-inducible factors HIF-1 and HIF-2, to adapt and survive [91,92]. It has been reported that PERK/eIF2α signaling plays a critical role in protecting chemotherapeutic resistant hypoxic cells, through induction of GSH synthesis, thus, reducing accumulation of ROS [93]. Combining hyperthermia with a low linear energy transfer (LET) radiation, could be the effective strategy to overcome hypoxia, to make cells sensitive to treatment, again [94,95].
PERK kinase and its downstream target, Nrf2, is the master transcriptional regulator of the cellular antioxidant and detoxifying systems, which are key mediators of chemotherapy resistance, in various tumor cells [96]. Nrf2 has been identified as a novel transcription substrate of PERK and is associated with Kelch-like ECH-associated protein 1 (Keap1), in unstressed cells, where activation of PERK via agents that trigger the ERSR is necessary and sufficient for dissociation of cytoplasmic Nrf2/Keap1 and a subsequent Nrf2 nuclear import [89,97]. A strong positive correlation between the Nrf2 levels and the resistance of three cancer cell lines has been shown for chemotherapeutic drugs, such as cisplatin, doxorubicin, and etoposide [98]. Nrf2 has been involved in the promotion of cell survival or chemotherapy resistance to cancer cells, following ERS [89]. Nrf2 is an attractive molecule as a therapeutic target in cancer. For example, ML385, a small molecule inhibitor of Nrf2, selectively alters therapeutic resistance in Keap1-deficient Non-small-cell lung carcinoma (NSCLC) tumors [99]. The ability of Nrf2 to activate cyto-protective detoxifying enzymes systems, including the cytochrome P450 (CYP) system, glutathione-S-transferase (GST), and uridine diphospho-glucuronosyltransferase (UGT), plays a crucial role in reducing ROS, leading to the prevention of apoptosis in cancer cells [100,101]. Nrf2 basically regulates the phase II detoxifying enzymes system, upon the exposure of cells to agents that promote the accumulation of ROS [102]. GSTs have been implicated in the detoxification of many chemotherapeutics. Elevated levels of GSTs have been associated with malignant transformation and drug resistance [103,104]. Overexpression of GST in cancer cells, enhances the detoxification of the anticancer drugs, leading to a reduction in the efficacy of cytotoxic damage to the cells [105]. Elevation of GST is also associated with resistance to apoptosis, through direct detoxification, by inhibition of the MAPK pathway [79,80]. Overexpression of GSTs ensures a high GSH concentration and increases GSH-transporters inside cancer cells, which are associated with high resistance to chemotherapeutic agents [106]. Several studies have suggested that a high GSH level is associated with an apoptotic resistance to the cancer cells [107,108]. The GSH metabolism has been reported to contribute to cancer cell resistance to anti-neoplastic treatments, either by inhibition of apoptosis, or through chemotherapeutic drug detoxification, by a GSH-conjugation [109]. The chemotherapeutic drugs are conjugated with GHS and efflux, through the GSH-pump of MRP transporters [110]. Nrf2-dependent proteins exert a pro-survival effect that can be through inducing HIF𝛼, increasing MRP or increasing the synthesis of GSH [111,112,113].
Multidrug resistance (MDR) is one of the main obstacles to treating cancer, as it can facilitate the efflux of anticancer drugs from cancer cells [64,65,114]. For example, PERK has been involved in the upregulation of MDR-related protein, MRP1, through the regulation of Nrf2 in the chemotherapy-resistant human colon adenocarcinoma cells line (HT29) [17]. In cancer cells, MDR is developed by upregulating antioxidant enzymes (hemeoxygenase 1, superoxide dismutase, catalase, etc.) that neutralize the ROS required for chemotherapy toxicity or by upregulating drug efflux pumps [101,115,116]. It has been reported that PERK-Nrf2 signaling protects de-differentiated cells from chemotherapy, by reducing ROS levels and increasing drug efflux, which suggests that targeting this pathway could sensitize drug-resistant cells to chemotherapy [97].
The ratio of anti-apoptotic Bcl-2 and apoptotic Bax proteins largely regulates cell death and survival [117,118,119]. It has been reported that Nrf2 exhibits protection against apoptosis, by increasing the levels of anti-apoptotic protein Bcl-2, in the hydrogen sulfide (H2S)-treated mice [120]. Elevated expression of Bcl-2 protein is associated with poor prognosis in many human cancers. It has been demonstrated that antioxidant exposure initiates Nrf2 stabilization, which leads to an increased Bcl-2/Bax ratio, subsequently reducing apoptosis and increasing cell survival [121]. The elevated expression of Bcl-2 protects lung cancer A549 cells from drug or radiation-induced DNA fragmentation and apoptosis [122,123].
It has been assumed that over-expression of Bcl-2 family members and the loss of wild-type p53 are mainly involved in resistance to apoptosis induced by chemotherapeutic drugs [124]. Bcl-2 constitutively controls p53-dependent apoptosis in HCT116 cellosaurus human colorectal cancer cells and A2780 ovarian cell line [125,126]. The tumor suppressor gene p53 plays an important role in chemotherapy resistance. Nrf2 regulates the basal expression of a direct inhibitor of p53, namely mouse double minute 2 homolog (Mdm2) [127]. The elevated expression of Nrf2 can cease ROS-based apoptotic signals, through the inhibition of p53, thus, enhancing cellular survival by leading to chemotherapy resistance [128]. It has also been reported that the p53/p21 complex regulates tumor cell invasion and apoptosis, by targeting Bcl-2 family proteins [129]. Nrf2 can inhibit the p53 target gene p21 by blocking interaction with Keap1 [130]. It has been found that Bcl-2 overexpression is associated with an increased resistance to cell death, after treatment with etoposide (DNA topoisomerase II inhibitor) [131]. The treatment of etoposide induces apoptosis, which is accompanied by the down-regulation of Bcl-2 expression, in small cell lung cancer (SCLC) and NSCLC cell lines [132].
The mammalian inhibitor of the apoptosis (IAP) gene family encoding proteins (XIAP, cIAP1 and cIAP2), contributes towards protecting the cells against a variety of apoptotic stimuli, resulting in drug resistance [133,134]. The cellular IAPs (cIAP1 and cIAP2) and the X-chromosome-linked IAP (XIAP) are highly expressed in many human malignancies [135,136,137,138,139]. It has been demonstrated that PERK activity can induce cellular inhibitor of apoptosis (cIAP1 and cIAP2) proteins, leading to the inhibition of ER stress-induced apoptotic program [140]. The ER stress inducing agents can activate the phosphatidylinositol 3-kinase (PI3K)–Akt signaling pathway, which is involved in the transcriptional induction of IAPs [141,142,143,144].
4.4.2. Role of Inositol-Requiring Enzyme 1 Alpha (IRE1), Activating Transcription Factor 6 Alpha (ATF6), and Glucose-Regulated Protein 78 (GRP78) in Chemotherapy Resistance
The inositol-requiring enzyme 1 alpha (IRE1), activating transcription factor 6 alpha (ATF6), and glucose-regulated protein 78 (GRP78), play a critical role in chemotherapy resistance in cancers (Figure 4).
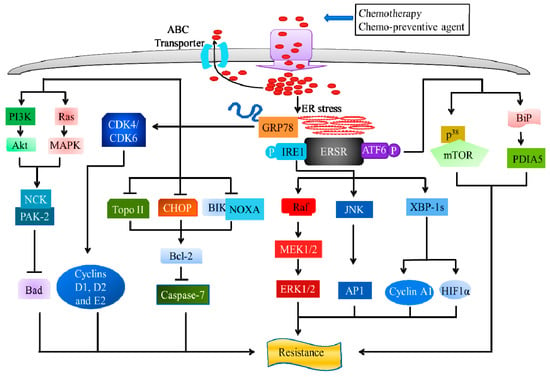
Figure 4.
ER stress and chemo-resistance: Role of inositol-requiring enzyme 1 alpha (IRE1), activating transcription factor 6 alpha (ATF6), and glucose-regulated protein 78 (GRP78). Upon ERS, IRE splices and activates the X-box-binding protein 1 (XBP1) that controls cycline A1 expression and regulates HIF1α targets. The activation of IRE1 leads to the activation of c-Jun N-terminal protein kinase (JNK), followed by inducing AP-1 transcriptional activity. The Raf/MEK/ERK pathway is also involved in IRE1-mediated chemo-resistance. ATF6α possesses chemo-resistance through the regulation of ER chaperone BiP protein, mediated by the activation of disulfide isomerase A5 (PDIA5) and p38-dependent ATF6α-mTOR pathway, upon ER stress. GRP78 exhibits its chemo-resistance properties, upon ERS, through the suppression of C/EBP homologous protein (CHOP), Bcl-2-interacting killer (BIK), phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) or NOXA, Topo II, and induction of Bcl-2, leading to the activation of caspase-7 dependent apoptosis. GRP78 suppresses G1/S transition-related cyclins (D1, E1, and E2) and cyclin-dependent kinase (CDK4 and CDK6) protein expression. Additionally, the phosphorylation of GRP78 leads to the activation of Ras/MAPK and PI3-kinase downstream signaling, and the recruitment of p21 activated kinases 2 (PAK-2) via NCK, thus, inhibiting the pro-apoptotic protein Bcl-2-associated death (Bad).
IRE1 could regulate cell survival induced by the ERS inducing agents, tunicamycin, and thapsigargin [145]. IRE1α, a subset of the ERSR, controls cyclin A1 expression and mediates proliferation, through tight control of XBP-1 splicing [146]. It has been established that XBP1 and HIF1α co-operatively regulate HIF1α targets, including vascular endothelial growth factor-A (VEGF-A), phosphoinositide-dependent kinase 1 (PDPK1), glucose transporter 1 (GLUT1), and DNA-damage-inducible transcript 4 (DDIT4), in triple-negative breast cancer (TNBC) [147].
Upon ERS, IRE1 recruits the tumor necrosis factor receptor (TNFR)-associated factor-2 (TRAF2) and activates apoptosis-signaling-kinase 1 (ASK1), leading to the activation of c-jun N-terminal protein kinase (JNK) [148,149]. ERSR can stimulate JNKs, which are important in controlling apoptosis in the process of cellular stress, thus, increasing the activator protein 1 (AP-1) transcriptional activity. It has been demonstrated that tamoxifen resistance is accompanied by an increased AP-1 DNA binding in MCF-7 cells [150].
The Raf/MEK/ERK pathway plays an important role in chemotherapeutic drug resistance, as activation of Raf induces resistance to doxorubicin and paclitaxel, in breast cancer cells [151,152]. The activation of extracellular signal-regulated kinase-1 and 2 (ERK1/2), is partially IRE1-dependent in azetidine-2 carboxylic acid-treated mouse embryonic fibroblasts (MEFs), in vitro [153]. Upon ER stress, the non-catalytic region of tyrosine kinase adaptor protein 1 (Nck1), dissociated from the ER membrane, increased activation of ERK1/2 and was correlated in vivo, with increased cell proliferation and reduced apoptosis [154].
ATF6 activation is essential for protecting melanoma cells against ERS-induced cell death [155]. Two Golgi resident enzymes, site-1 protease (S1P) and site-2 protease (S2P), are involved in the proteolytic cleavage of the full-length 90-kDa ATF6 [156,157]. It has been identified that Notch1 acts as a novel transcriptional target of ATF6, with a potential role in promoting an anti-apoptotic phenotype in irradiated glioblastoma [158]. The induction of ATF6α was required for cell survival and the expression of ER chaperones, upon ERS in the ATF6α knockout mouse embryonic fibroblasts (MEFs) [159,160]. The expression of the ER chaperone BiP protects the cells from ERS [161]. A novel role for ATF6α in chemo-resistance through the protein disulfide isomerase A5 (PDIA5) upon ER stress has been identified, in which genetic and pharmacological inhibition of the PDIA5/ATF6α, could restore sensitivity to the chemotherapy treatment [162]. The transcription factor ATF6α has been identified as a pivotal survival factor for quiescent squamous carcinoma cells [163]. Knockdown of ATF6α prolonged the survival of nude mice bearing dormant tumor cells through the ATF6α-Rheb-mammalian target of rapamycin (mTOR) pathway in a p38-dependent manner.
GRP78, also referred to as BiP, is a major molecular chaperone protein with anti-apoptotic properties, which plays a key role as a pro-survival component of ERSR [164,165,166]. The level of GRP78 is elevated in a variety of cancer cell lines and solid tumors associated with malignancy, and correlates with cellular drug resistance [167,168,169,170,171]. It has been demonstrated that under-expression of GRP78, induces apoptosis of colon cancer cells, in vitro, and suppresses tumor formation of colon cancer cells in a mouse xenograft model [172]. Enhanced pro-survival signaling resulting from ERSR has been found to promote resistance to chemotherapy, through upregulation of the canonical targets, such as GRP78 [173]. The induction of GRP78 could lead to cancer progression and drug resistance in neoplastic cells. On the other hand, overexpression of GRP78 could limit damage in normal tissues and organs exposed to ERS, due to its anti-apoptotic function [164]. Overexpression of GRP78 induced by celecoxib, partially suppresses the induction of CHOP and protects cancer cells from celecoxib-induced apoptosis in human GC cells [174]. It has been revealed that overexpression of GRP78, suppresses apoptosis induced by the Bcl-2-interacting killer (BIK) and NOXA, leading to activation of Bcl-2 and blocking of caspase-dependent apoptosis [175,176]. It has been reported that GRP78 protects cells from apoptosis induced by Topo II inhibitors (etoposide), by blocking the activation of caspase-7, both in vivo and in vitro [176,177]. The evidence suggests that mutation or altered expression of the Topo II genes are sufficient to confer resistance to topoisomerase inhibitors [178]. The under-expression of Topo II in tumor cells, due to mutations and posttranslational modification, leads to a reduction in the formation of the cleavable-complex in cancer cells, thus, preventing apoptosis of tumor cells [179,180]. Several studies have demonstrated that ERSR leads to a decrease in Topo II α protein and a concomitant resistance to chemotherapeutic agents that target this enzyme [168,181]. It has been reported that ERSR is essential and sufficient for reducing Topo IIα protein levels, thereby, increasing the resistance to topoisomerase-targeted drugs, like etoposide and doxorubicin [182]. The glucose-regulated proteins (GPRs) actively reduced Topo II expression in isolated nuclei from K12 cells, during a stress response, which is responsible for cellular resistance to etoposide [181,183].
The anticancer drugs 5-fluorouracil (5-FU) and doxorubicin become desensitized to renal cell carcinoma (RCC), due to the silencing of GRP78, by small interfering ribonucleic acid (siRNA) that induced G1 cell-cycle arrest and inhibited the growth of cells [184]. The specific inhibition of GRP78 expression suppressed G1/S transition-related cyclins (D1, E1, and E2) and cyclin-dependent kinase (CDK4 and CDK6) protein expression [184].
A study has demonstrated that GRP78 secreted by colon cancer cells facilitates cell proliferation via the PI3K/Akt signaling. It also showed that PI3K/Akt inhibition promoted ERS-induced apoptosis in a GRP78-dependent manner [185]. GRP78 contributes to the proliferation and tumorigenesis of human colorectal carcinoma, via the Akt and MAPK pathways [186]. An emerging study indicated that binding of activated alpha2-macroglobulin (α2-macroglobulin) to surface GRP78 activates p21-activated kinases (PAKs)-dependent signaling and promotes proliferation and metastasis in 1-LN prostate cancer cells [187]. The phosphorylation of GRP78, leads to the activation of Ras/MAPK and PI3-kinase downstream signaling, and recruitment of PAK-2, via the adaptor protein, NCK to the plasma membrane, thus, inhibiting the pro-apoptotic protein Bcl-2-associated death (Bad) [183].
5. Conclusions
Chemotherapy resistance is the tolerance of tumor cells to cancer treatments, which is a significant issue in the management of cancer. As such, chemotherapy resistance can be a major barrier for the physician in the treatment of cancer. Only a thorough understanding of the mechanisms of chemotherapy resistance will allow the physician to set the therapy as needed. While the development of new chemotherapies has proceeded quickly, chemotherapeutic agents that are effective against the advanced stages of cancer (such as invasion and metastasis) have not been discovered, due to a poor understanding of chemotherapy resistance. The ERSR has a vital role in the adaptive survival signaling that promotes resistance to chemotherapy in cancer cells. The chemotherapy resistance resulting from the ERSR is more or less regulated by three ER-resident sensor proteins, namely PERK, ATF6, and IRE1α. In these circumstances, ERSR could become a potential and effective target for the new development of chemotherapeutics. However, the detailed molecular mechanisms of the relationship between the ERSR and chemotherapy resistance remain poorly understand. In this review, we explained the molecular mechanism of ER stress-mediated apoptosis on cancer treatments, and also correlated the most remarkable pathways of the interplay between the ERSR and chemotherapy resistance.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar]
- Heron, M.; Anderson, R.N. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality; NCHS Data Brief 2016; NCHS: Hyattsville, MD, USA, 2016; pp. 1–8. [Google Scholar]
- Korean Statistical Information Service National Cancer Registration Data. Available online: www.kosis.kr (accessed on 6 July 2018).
- Chakraborty, S.; Rahman, T. The difficulties in cancer treatment. Ecancermedicalscience 2012, 6, ed16. [Google Scholar] [PubMed]
- Liu, B.; Ezeogu, L.; Zellmer, L.; Yu, B.; Xu, N.; Joshua Liao, D. Protecting the normal in order to better kill the cancer. Cancer Med. 2015, 4, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Norouzi-Barough, L.; Sarookhani, M.R.; Sharifi, M.; Moghbelinejad, S.; Jangjoo, S.; Salehi, R. Molecular mechanisms of drug resistance in ovarian cancer. J. Cell. Physiol. 2018, 233, 4546–4562. [Google Scholar] [CrossRef] [PubMed]
- Tesarova, P. Specific Aspects of Breast Cancer Therapy of Elderly Women. Biomed Res. Int. 2016, 2016, 1381695. [Google Scholar] [CrossRef] [PubMed]
- Henson, K.E.; Fry, A.; Lyratzopoulos, G.; Peake, M.; Roberts, K.J.; McPhail, S. Sociodemographic variation in the use of chemotherapy and radiotherapy in patients with stage IV lung, oesophageal, stomach and pancreatic cancer: Evidence from population-based data in England during 2013–2014. Br. J. Cancer 2018, 118, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Arruebo, M.; Vilaboa, N.; Saez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; Gonzalez-Fernandez, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef]
- Raguz, S.; Yague, E. Resistance to chemotherapy: New treatments and novel insights into an old problem. Br. J. Cancer 2008, 99, 387–391. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. CMLS 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. Endoplasmic reticulum stress response and transcriptional reprogramming. Front. Genet. 2014, 5, 460. [Google Scholar] [PubMed]
- Healy, S.J.; Gorman, A.M.; Mousavi-Shafaei, P.; Gupta, S.; Samali, A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharmacol. 2009, 625, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Yu, X.S.; Du, J.; Fan, Y.J.; Liu, F.J.; Cao, L.L.; Liang, N.; Xu, D.G.; Zhang, J.D. Activation of endoplasmic reticulum stress promotes autophagy and apoptosis and reverses chemo-resistance of human small cell lung cancer cells by inhibiting the PI3K/AKT/mTOR signaling pathway. Oncotarget 2016, 7, 76827–76839. [Google Scholar] [CrossRef]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Makin, G.; Dive, C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001, 11, S22–S26. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A. Targeting the extrinsic apoptotic pathway in cancer: Lessons learned and future directions. J. Clin. Investig. 2015, 125, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L. Shaping the dynamic mitochondrial network. BMC Biol. 2014, 12, 35. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Wongrakpanich, A.; Geary, S.M.; Joiner, M.L.; Anderson, M.E.; Salem, A.K. Mitochondria-targeting particles. Nanomedicine 2014, 9, 2531–2543. [Google Scholar] [CrossRef]
- Modica-Napolitano, J.S.; Weissig, V. Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 17394–17421. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Arbabian, A.; Brouland, J.P.; Gelebart, P.; Kovacs, T.; Bobe, R.; Enouf, J.; Papp, B. Endoplasmic reticulum calcium pumps and cancer. BioFactors 2011, 37, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Targeting endoplasmic reticulum signaling pathways in cancer. Acta Oncol. 2012, 51, 822–830. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Liu, F.G.; Wei, C.F.; Lu, C.C.; Chen, C.C.; Lin, H.C.; Ojcius, D.M.; Lai, H.C. Activation of multiple apoptotic pathways in human nasopharyngeal carcinoma cells by the prenylated isoflavone, osajin. PLoS ONE 2011, 6, e18308. [Google Scholar] [CrossRef] [PubMed]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Pan, M.Y.; Shen, Y.C.; Lu, C.H.; Yang, S.Y.; Ho, T.F.; Peng, Y.T.; Chang, C.C. Prodigiosin activates endoplasmic reticulum stress cell death pathway in human breast carcinoma cell lines. Toxicol. Appl. Pharmacol. 2012, 265, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Kurathong, S.; Punyagupta, S.; Lolekha, S.; Jayanetra, P. Anaerobic infections: Clinical, bacteriological and therapeutic features of thirty-six patients. J. Med Assoc. Thail. 1975, 58, 125–137. [Google Scholar]
- Hedgepeth, S.C.; Garcia, M.I.; Wagner, L.E., 2nd; Rodriguez, A.M.; Chintapalli, S.V.; Snyder, R.R.; Hankins, G.D.; Henderson, B.R.; Brodie, K.M.; Yule, D.I.; et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J. Biol. Chem. 2015, 290, 7304–7313. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Hosono, M.; Miura, Y.; Sugawara, S.; Kariya, Y.; Hakomori, S.; Nitta, K. Involvement of ER stress in apoptosis induced by sialic acid-binding lectin (leczyme) from bullfrog eggs. Int. J. Oncol. 2013, 43, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- White, N. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1999, 354, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef]
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Tamai, I.; Takaki, A.; Tsuji, A. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int. J. Cancer 2000, 88, 274–280. [Google Scholar] [CrossRef]
- Keppler, D. Multidrug resistance proteins (MRPs, ABCCs): Importance for pathophysiology and drug therapy. Handb. Exp. Pharmacol. 2011, 299–323. [Google Scholar] [CrossRef]
- Garcia-Carrasco, M.; Mendoza-Pinto, C.; Macias Diaz, S.; Vera-Recabarren, M.; Vazquez de Lara, L.; Mendez Martinez, S.; Soto-Santillan, P.; Gonzalez-Ramirez, R.; Ruiz-Arguelles, A. P-glycoprotein in autoimmune rheumatic diseases. Autoimmun. Rev. 2015, 14, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, H.; Yang, K.; Xu, W.; Tang, W.; Li, X. The structure and functions of P-glycoprotein. Curr. Med. Chem. 2010, 17, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Bikadi, Z.; Rosenberg, M.F.; Mao, Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr. Drug Metab. 2010, 11, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Riha, J.; Wlcek, K.; Jaeger, W.; Thalhammer, T. Organic anion transporting polypeptides (OATPs): Regulation of expression and function. Curr. Drug Metab. 2011, 12, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Gulati, A.; Gerk, P.M. Role of placental ATP-binding cassette (ABC) transporters in antiretroviral therapy during pregnancy. J. Pharm. Sci. 2009, 98, 2317–2335. [Google Scholar] [CrossRef]
- Golin, J.; Ambudkar, S.V. The multidrug transporter Pdr5 on the 25th anniversary of its discovery: An important model for the study of asymmetric ABC transporters. Biochem. J. 2015, 467, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qiu, M.; Tang, Q.L.; Liu, M.; Lang, N.; Bi, F. Establishment and biological characteristics of oxaliplatin-resistant human colon cancer cell lines. Chin. J. Cancer 2010, 29, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, L.; Kjellstrom, J.; Johnsson, A. Reduced drug accumulation is more important in acquired resistance against oxaliplatin than against cisplatin in isogenic colon cancer cells. Anticancer Drugs 2010, 21, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Meijer, G.A.; Schroeijers, A.B.; Flens, M.J.; Meuwissen, S.G.; van der Valk, P.; Baak, J.P.; Scheper, R.J. Increased expression of multidrug resistance related proteins Pgp, MRP1, and LRP/MVP occurs early in colorectal carcinogenesis. J. Clin. Pathol. 1999, 52, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Klappe, K.; Hinrichs, J.W.; Kroesen, B.J.; Sietsma, H.; Kok, J.W. MRP1 and glucosylceramide are coordinately over expressed and enriched in rafts during multidrug resistance acquisition in colon cancer cells. Int. J. Cancer 2004, 110, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F. DNA topoisomerase poisons as antitumor drugs. Annu. Rev. Biochem. 1989, 58, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- Stavrovskaya, A.A. Cellular mechanisms of multidrug resistance of tumor cells. Biochem. Biokhimiia 2000, 65, 95–106. [Google Scholar]
- Hinds, M.; Deisseroth, K.; Mayes, J.; Altschuler, E.; Jansen, R.; Ledley, F.D.; Zwelling, L.A. Identification of a point mutation in the topoisomerase II gene from a human leukemia cell line containing an amsacrine-resistant form of topoisomerase II. Cancer Res. 1991, 51, 4729–4731. [Google Scholar] [PubMed]
- Zwelling, L.A.; Hinds, M.; Chan, D.; Mayes, J.; Sie, K.L.; Parker, E.; Silberman, L.; Radcliffe, A.; Beran, M.; Blick, M. Characterization of an amsacrine-resistant line of human leukemia cells. Evidence for a drug-resistant form of topoisomerase II. J. Biol. Chem. 1989, 264, 16411–16420. [Google Scholar] [PubMed]
- Hait, W.N.; Choudhury, S.; Srimatkandada, S.; Murren, J.R. Sensitivity of K562 human chronic myelogenous leukemia blast cells transfected with a human multidrug resistance cDNA to cytotoxic drugs and differentiating agents. J. Clin. Investig. 1993, 91, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmoller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Novotna, R.; Wsol, V.; Xiong, G.; Maser, E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol. Lett. 2008, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharm. Rev 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, A.; Verma, A.; Mehta, K. Tissue transglutaminase promotes or suppresses tumors depending on cell context. Anticancer Res 2009, 29, 1909–1919. [Google Scholar] [PubMed]
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194. [Google Scholar] [CrossRef]
- Cumming, R.C.; Lightfoot, J.; Beard, K.; Youssoufian, H.; O’Brien, P.J.; Buchwald, M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat. Med. 2001, 7, 814–820. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anticancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Haeri, M.; Knox, B.E. Endoplasmic Reticulum Stress and Unfolded Protein Response Pathways: Potential for Treating Age-related Retinal Degeneration. J. Ophthalmic Vis. Res. 2012, 7, 45–59. [Google Scholar] [PubMed]
- Dufey, E.; Sepulveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Fels, D.R.; Koumenis, C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol. Ther. 2006, 5, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Surviving ischemia: Adaptive responses mediated by hypoxia-inducible factor 1. J. Clin. Investig. 2000, 106, 809–812. [Google Scholar] [CrossRef]
- Ratcliffe, P.J.; O’Rourke, J.F.; Maxwell, P.H.; Pugh, C.W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 1998, 201, 1153–1162. [Google Scholar]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Elming, P.B.; Sorensen, B.S.; Oei, A.L.; Franken, N.A.P.; Crezee, J.; Overgaard, J.; Horsman, M.R. Hyperthermia: The Optimal Treatment to Overcome Radiation Resistant Hypoxia. Cancers 2019, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.; Krawczyk, P.M.; Horsman, M.R.; Franken, N.A.; Crezee, J. Targeting therapy-resistant cancer stem cells by hyperthermia. Int. J. Hyperth. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001, 174, 103–113. [Google Scholar] [CrossRef]
- Cazenave, L.A.; Moscow, J.A.; Myers, C.E.; Cowan, K.H. Glutathione S-transferase and drug resistance. Cancer Treat. Res. 1989, 48, 171–187. [Google Scholar] [PubMed]
- Ramsay, E.E.; Dilda, P.J. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front. Pharmacol. 2014, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Manolitsas, T.P.; Englefield, P.; Eccles, D.M.; Campbell, I.G. No association of a 306-bp insertion polymorphism in the progesterone receptor gene with ovarian and breast cancer. Br. J. Cancer 1997, 75, 1398–1399. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Ebbert, J.O.; Sun, Z.; Weinshilboum, R.M. Role of the glutathione metabolic pathway in lung cancer treatment and prognosis: A review. J. Clin. Oncol. 2006, 24, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.; Berson, A.; Haouzi, D.; Vadrot, N.; Fau, D.; Grodet, A.; Letteron, P.; Feldmann, G.; El-Benna, J.; Fromenty, B.; et al. High hepatic glutathione stores alleviate Fas-induced apoptosis in mice. J. Hepatol. 2007, 46, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.J.; Pappa, A.; Panayiotidis, M.I. The central role of glutathione in the pathophysiology of human diseases. Arch. Physiol. Biochem. 2007, 113, 234–258. [Google Scholar] [CrossRef]
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Y.; Xia, S.; Rao, B.; Zhang, J.; Wang, J. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 2014, 27, 685–692. [Google Scholar] [CrossRef]
- Li, C.; Wu, H.; Wang, S.; Zhu, J. Expression and correlation of NRF2, KEAP1, NQO-1 and HO-1 in advanced squamous cell carcinoma of the larynx and their association with clinicopathologic features. Mol. Med. Rep. 2016, 14, 5171–5179. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. How cancer cells evade chemotherapy: Sixteenth Richard and Hinda Rosenthal Foundation Award Lecture. Cancer Res. 1993, 53, 747–754. [Google Scholar] [PubMed]
- Simon, S.M.; Schindler, M. Cell biological mechanisms of multidrug resistance in tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Aravind, L.; Dixit, V.M.; Koonin, E.V. Apoptotic molecular machinery: Vastly increased complexity in vertebrates revealed by genome comparisons. Science 2001, 291, 1279–1284. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Li, Q.K.; Singh, A.; Biswal, S.; Askin, F.; Gabrielson, E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J. Hum. Genet. 2011, 56, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.A.; Li, G. Cancer chemoresistance: The relationship between p53 and multidrug transporters. Int. J. Cancer 2002, 98, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Milner, J. Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 2003, 17, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Kerr, D.J.; Herod, J.; Hodgkins, L.; Krajewski, S.; Reed, J.C.; Young, L.S. The control of apoptosis and drug resistance in ovarian cancer: Influence of p53 and Bcl-2. Oncogene 1995, 11, 1217–1228. [Google Scholar] [PubMed]
- You, A.; Nam, C.W.; Wakabayashi, N.; Yamamoto, M.; Kensler, T.W.; Kwak, M.K. Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch. Biochem. Biophys. 2011, 507, 356–364. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef]
- Kim, E.M.; Jung, C.H.; Kim, J.; Hwang, S.G.; Park, J.K.; Um, H.D. The p53/p21 Complex Regulates Cancer Cell Invasion and Apoptosis by Targeting Bcl-2 Family Proteins. Cancer Res. 2017, 77, 3092–3100. [Google Scholar] [CrossRef]
- Rotblat, B.; Melino, G.; Knight, R.A. NRF2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272–1283. [Google Scholar] [CrossRef]
- Yoshida, A.; Takemura, H.; Inoue, H.; Miyashita, T.; Ueda, T. Inhibition of glutathione synthesis overcomes Bcl-2-mediated topoisomerase inhibitor resistance and induces nonapoptotic cell death via mitochondrial-independent pathway. Cancer Res. 2006, 66, 5772–5780. [Google Scholar] [CrossRef]
- Oizumi, S.; Isobe, H.; Ogura, S.; Ishida, T.; Yamazaki, K.; Nishimura, M.; Kawakami, Y.; Dosaka-Akita, H. Topoisomerase inhibitor-induced apoptosis accompanied by down-regulation of Bcl-2 in human lung cancer cells. Anticancer Res. 2002, 22, 4029–4037. [Google Scholar]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [PubMed]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D.; Fairbrother, W.J. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin. Cancer Res. 2007, 13, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J.; Sheu, T.T.; Richter, B.W.; He, W.W.; Thornberry, N.A.; Duckett, C.S.; Hardwick, J.M. C-IAP1 is cleaved by caspases to produce a proapoptotic C-terminal fragment. J. Biol. Chem. 2001, 276, 7602–7608. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Beinroth, S.; Gleichmann, M.; Liston, P.; Korneluk, R.G.; MacKenzie, A.E.; Bahr, M.; Klockgether, T.; Robertson, G.S.; Weller, M.; et al. Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J. Neurochem. 1999, 72, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997, 16, 6914–6925. [Google Scholar] [CrossRef] [PubMed]
- Orth, K.; Dixit, V.M. Bik and Bak induce apoptosis downstream of CrmA but upstream of inhibitor of apoptosis. J. Biol. Chem. 1997, 272, 8841–8844. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Ji, X.; Liebhaber, S.A.; Diehl, J.A. PERK-dependent regulation of IAP translation during ER stress. Oncogene 2009, 28, 910–920. [Google Scholar] [CrossRef]
- Hosoi, T.; Hyoda, K.; Okuma, Y.; Nomura, Y.; Ozawa, K. Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 2007, 1152, 27–31. [Google Scholar] [CrossRef]
- Medcalf, G.W. The effect of school dental care on caries, oral hygiene, gingivitis, and calculus in western Australian children. Aust. Dent. J. 1983, 28, 239–242. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, S.; Mounir, Z.; Baltzis, D.; Raven, J.F.; Wang, S.; Krishnamoorthy, J.L.; Pluquet, O.; Pelletier, J.; Koromilas, A.E. A novel function of eIF2alpha kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol. Biol. Cell 2007, 18, 3635–3644. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.A.; Schwarze, S.R. IRE1alpha controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones 2010, 15, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Lewis-Wambi, J.S.; Jordan, V.C. Estrogen regulation of apoptosis: How can one hormone stimulate and inhibit? Breast Cancer Res. BCR 2009, 11, 206. [Google Scholar] [CrossRef]
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.; Chappell, W.H.; Basecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef]
- Weinstein-Oppenheimer, C.R.; Henriquez-Roldan, C.F.; Davis, J.M.; Navolanic, P.M.; Saleh, O.A.; Steelman, L.S.; Franklin, R.A.; Robinson, P.J.; McMahon, M.; McCubrey, J.A. Role of the Raf signal transduction cascade in the in vitro resistance to the anticancer drug doxorubicin. Clin. Cancer Res. 2001, 7, 2898–2907. [Google Scholar]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Kebache, S.; Fazel, A.; Wong, H.N.; Jenna, S.; Emadali, A.; Lee, E.H.; Bergeron, J.J.; Kaufman, R.J.; Larose, L.; et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 2004, 15, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Zhang, S.X.; Ma, J.H.; Bhatta, M.; Fliesler, S.J.; Wang, J.J. The unfolded protein response in retinal vascular diseases: Implications and therapeutic potential beyond protein folding. Prog. Retin. Eye Res. 2015, 45, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Fassl, A.; Tagscherer, K.E.; Richter, J.; Berriel Diaz, M.; Alcantara Llaguno, S.R.; Campos, B.; Kopitz, J.; Herold-Mende, C.; Herzig, S.; Schmidt, M.H.; et al. Notch1 signaling promotes survival of glioblastoma cells via EGFR-mediated induction of anti-apoptotic Mcl-1. Oncogene 2012, 31, 4698–4708. [Google Scholar] [CrossRef] [PubMed]
- Tay, K.H.; Luan, Q.; Croft, A.; Jiang, C.C.; Jin, L.; Zhang, X.D.; Tseng, H.Y. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal. 2014, 26, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Morris, J.A.; Dorner, A.J.; Edwards, C.A.; Hendershot, L.M.; Kaufman, R.J. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J. Biol. Chem. 1997, 272, 4327–4334. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. N. Y. 2004, 71, 289–297. [Google Scholar]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, S.; Liu, J.; Wang, X.; Ji, J.; Cao, Y.; Lu, K.; Wang, J.; Gao, Y. Expression of GRP78 predicts taxane-based therapeutic resistance and recurrence of human gastric cancer. Exp. Mol. Pathol. 2014, 96, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hughes, C.; Chao, C.; Cai, J.; Bartels, C.; Gessner, T.; Subjeck, J. Coinduction of glucose-regulated proteins and doxorubicin resistance in Chinese hamster cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3278–3282. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Miele, C.; Di Jeso, B.; Beguinot, F.; Nappi, C.; Di Carlo, C.; Capuozzo, S.; Terrazzano, G.; Insabato, L.; Ulianich, L. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol. Oncol. 2012, 125, 220–225. [Google Scholar] [CrossRef]
- Koomagi, R.; Mattern, J.; Volm, M. Glucose-related protein (GRP78) and its relationship to the drug-resistance proteins P170, GST-pi, LRP56 and angiogenesis in non-small cell lung carcinomas. Anticancer Res. 1999, 19, 4333–4336. [Google Scholar]
- Tomida, A.; Tsuruo, T. Drug resistance mediated by cellular stress response to the microenvironment of solid tumors. Anticancer Drug Des. 1999, 14, 169–177. [Google Scholar]
- Kosakowska-Cholody, T.; Lin, J.; Srideshikan, S.M.; Scheffer, L.; Tarasova, N.I.; Acharya, J.K. HKH40A downregulates GRP78/BiP expression in cancer cells. Cell Death Dis. 2014, 5, e1240. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, S.; Namba, T.; Tanaka, K.I.; Arai, Y.; Ishihara, T.; Aburaya, M.; Mima, S.; Hoshino, T.; Mizushima, T. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene 2006, 25, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Fu, Y.; Chan, L.; Lee, A.S. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): Endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK). J. Biol. Chem. 2011, 286, 25687–25696. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Beck, W.T.; Morgan, S.E.; Mo, Y.Y.; Bhat, U.G. Tumor cell resistance to DNA topoisomerase II inhibitors: New developments. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 1999, 2, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.E. Apoptosis in cancer therapy: Crossing the threshold. Cell 1994, 78, 539–542. [Google Scholar] [CrossRef]
- Matsuo, K.; Kohno, K.; Takano, H.; Sato, S.; Kiue, A.; Kuwano, M. Reduction of drug accumulation and DNA topoisomerase II activity in acquired teniposide-resistant human cancer KB cell lines. Cancer Res. 1990, 50, 5819–5824. [Google Scholar]
- Yun, J.; Tomida, A.; Nagata, K.; Tsuruo, T. Glucose-regulated stresses confer resistance to VP-16 in human cancer cells through a decreased expression of DNA topoisomerase II. Oncol. Res. 1995, 7, 583–590. [Google Scholar]
- Gray, M.D.; Mann, M.; Nitiss, J.L.; Hendershot, L.M. Activation of the unfolded protein response is necessary and sufficient for reducing topoisomerase IIalpha protein levels and decreasing sensitivity to topoisomerase-targeted drugs. Mol. Pharmacol. 2005, 68, 1699–1707. [Google Scholar]
- Shen, J.W.; Subjeck, J.R.; Lock, R.B.; Ross, W.E. Depletion of topoisomerase II in isolated nuclei during a glucose-regulated stress response. Mol. Cell. Biol. 1989, 9, 3284–3291. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.A.; Fang, S.U.; Su, C.L.; Hsiao, C.J.; Chang, C.C.; Lin, Y.F.; Cheng, C.W. Silencing glucose-regulated protein 78 induced renal cell carcinoma cell line G1 cell-cycle arrest and resistance to conventional chemotherapy. Urol. Oncol. 2014, 32, 29e21-11. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.Y.; Chen, S.K.; Yan, D.M.; Chen, R.; Lui, Y.P.; Duan, C.Y.; Li, J.; He, T.; Li, H. PI3K/Akt promotes GRP78 accumulation and inhibits endoplasmic reticulum stress-induced apoptosis in HEK293 cells. Folia Biol. 2010, 56, 37–46. [Google Scholar]
- Wang, Y.; Wang, J.H.; Zhang, X.L.; Wang, X.L.; Yang, L. Endoplasmic reticulum chaperone glucose-regulated protein 78 in gastric cancer: An emerging biomarker. Oncol. Lett. 2018, 15, 6087–6093. [Google Scholar] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).