Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
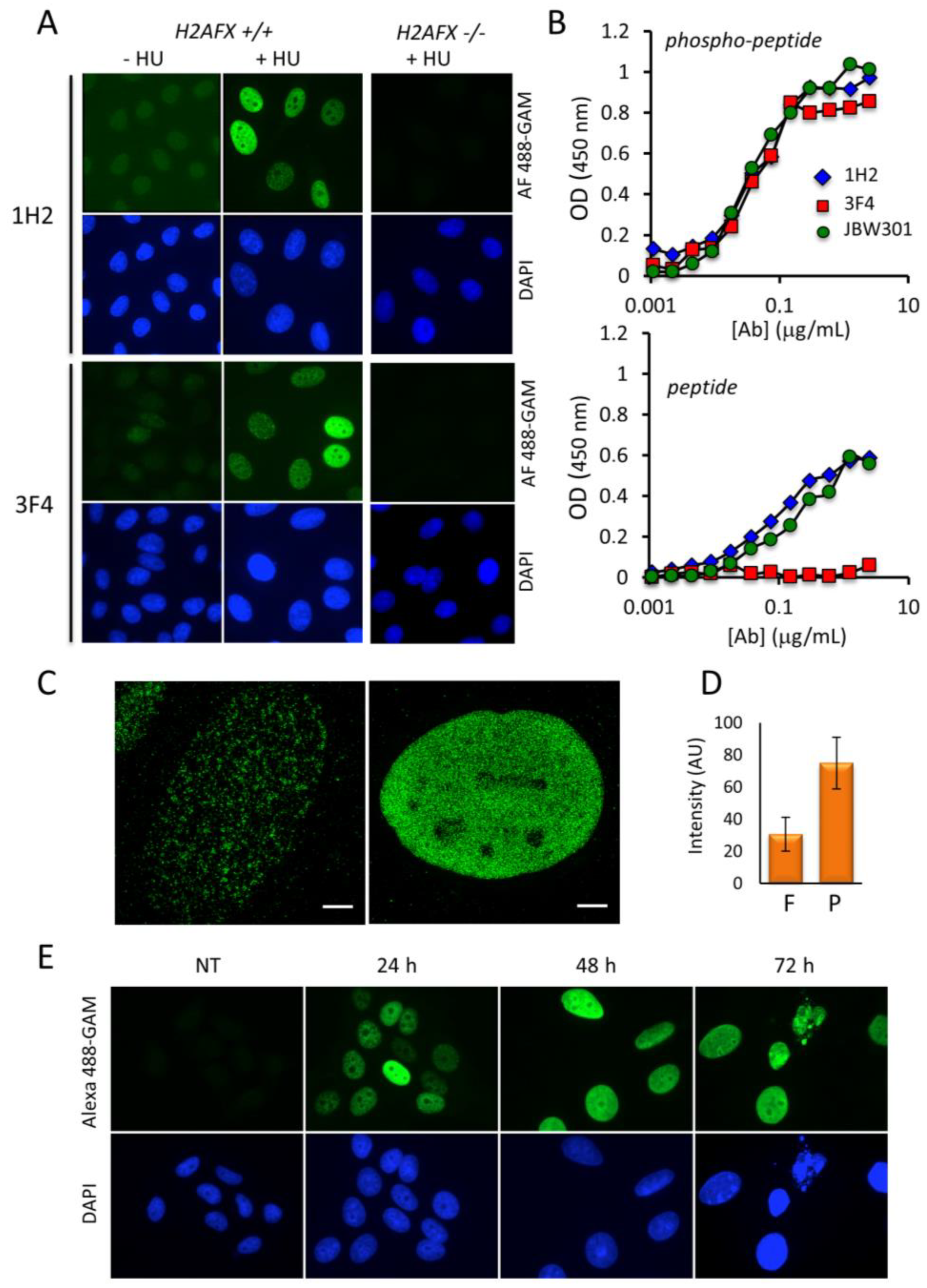
2.1. Analysis of the Dynamics of Phosphorylated H2AX with Newly-Generated Monoclonal Antibodies
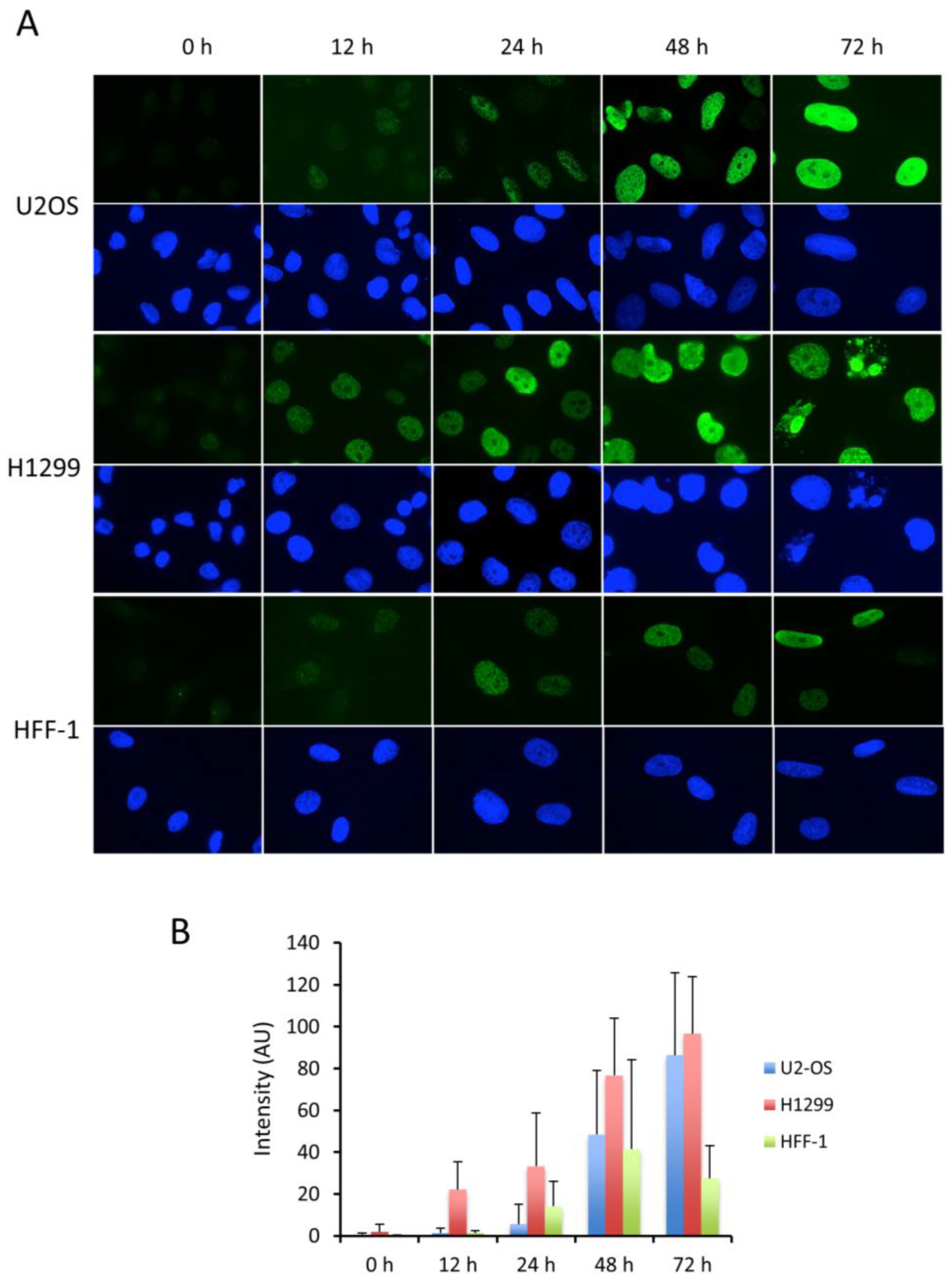
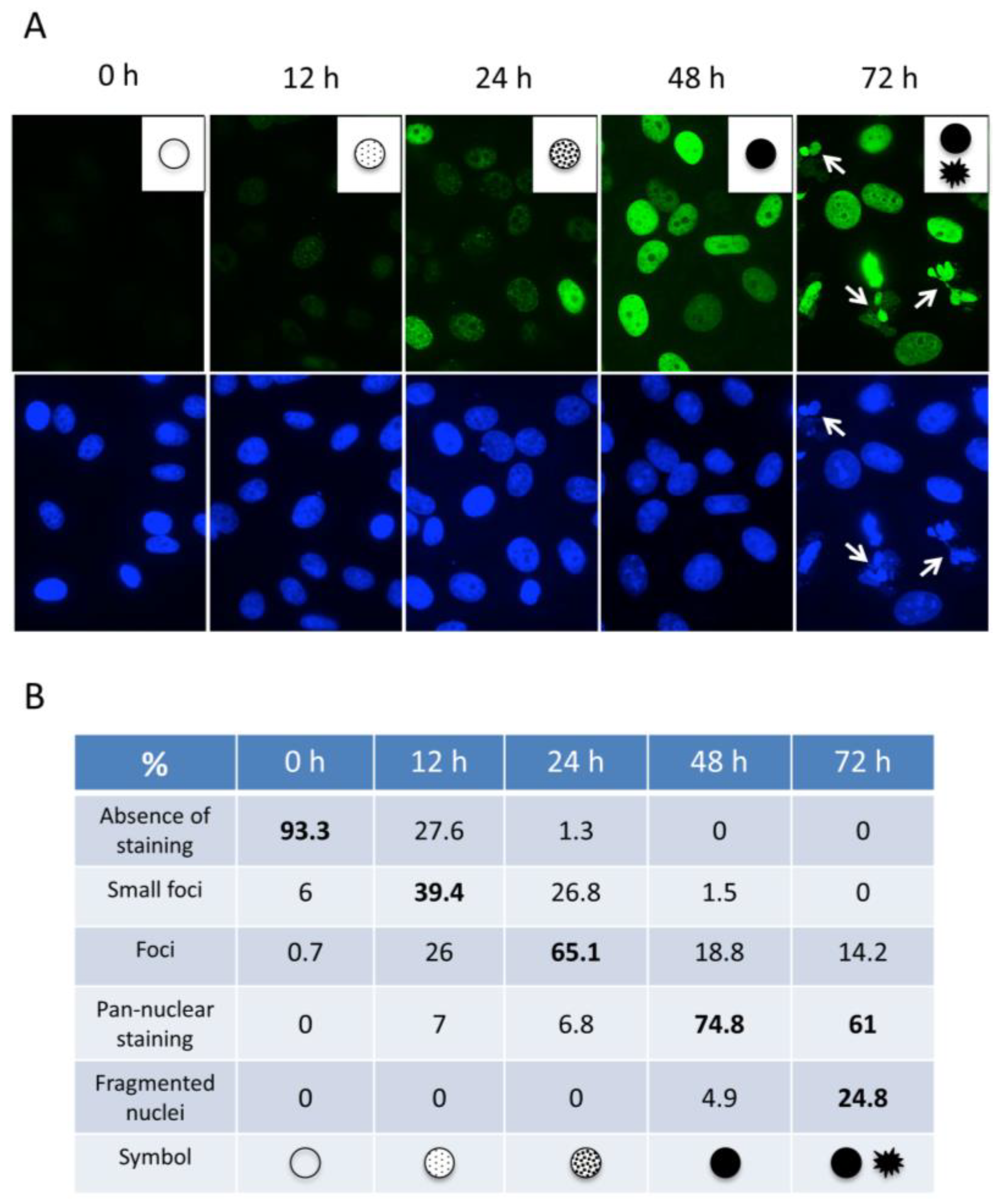
2.2. The Appearance of the Pan-Nuclear γ-H2AX Pattern Is Time and Drug-Dependent
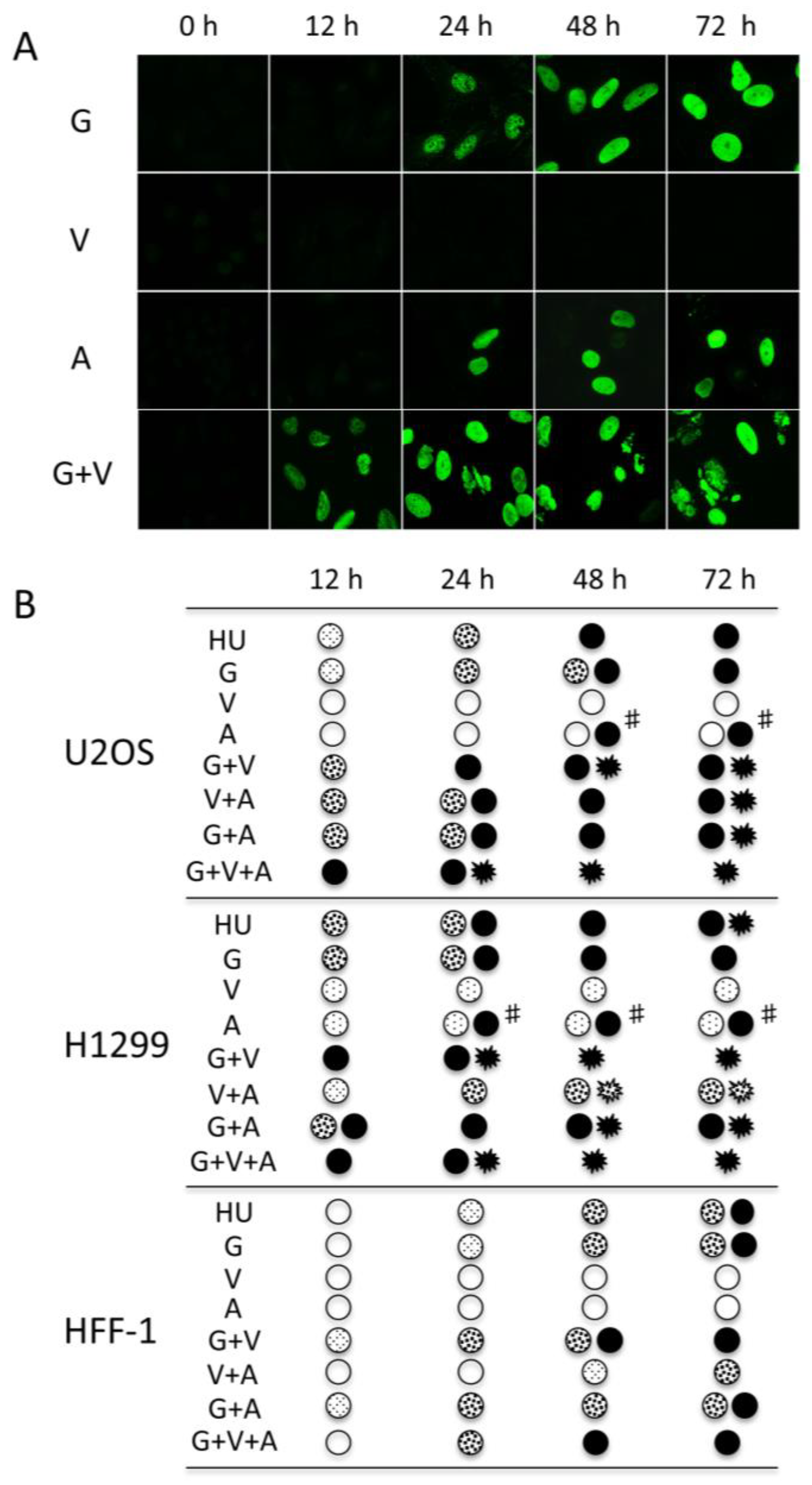
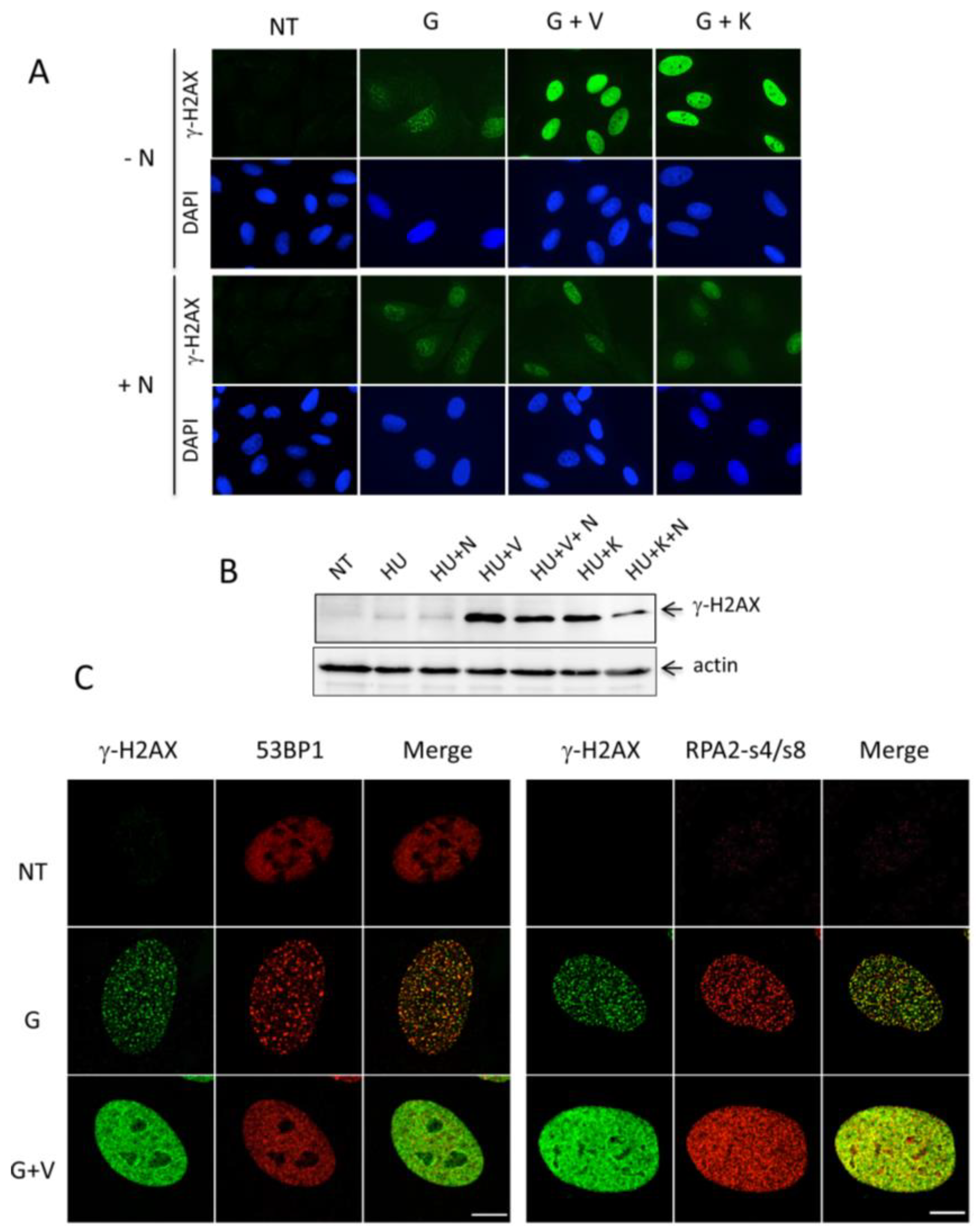
2.3. The Accumulation of the Widespread Pan-Nuclear γ-H2AX Pattern Is a Result of DNA-PK Hyperactivation
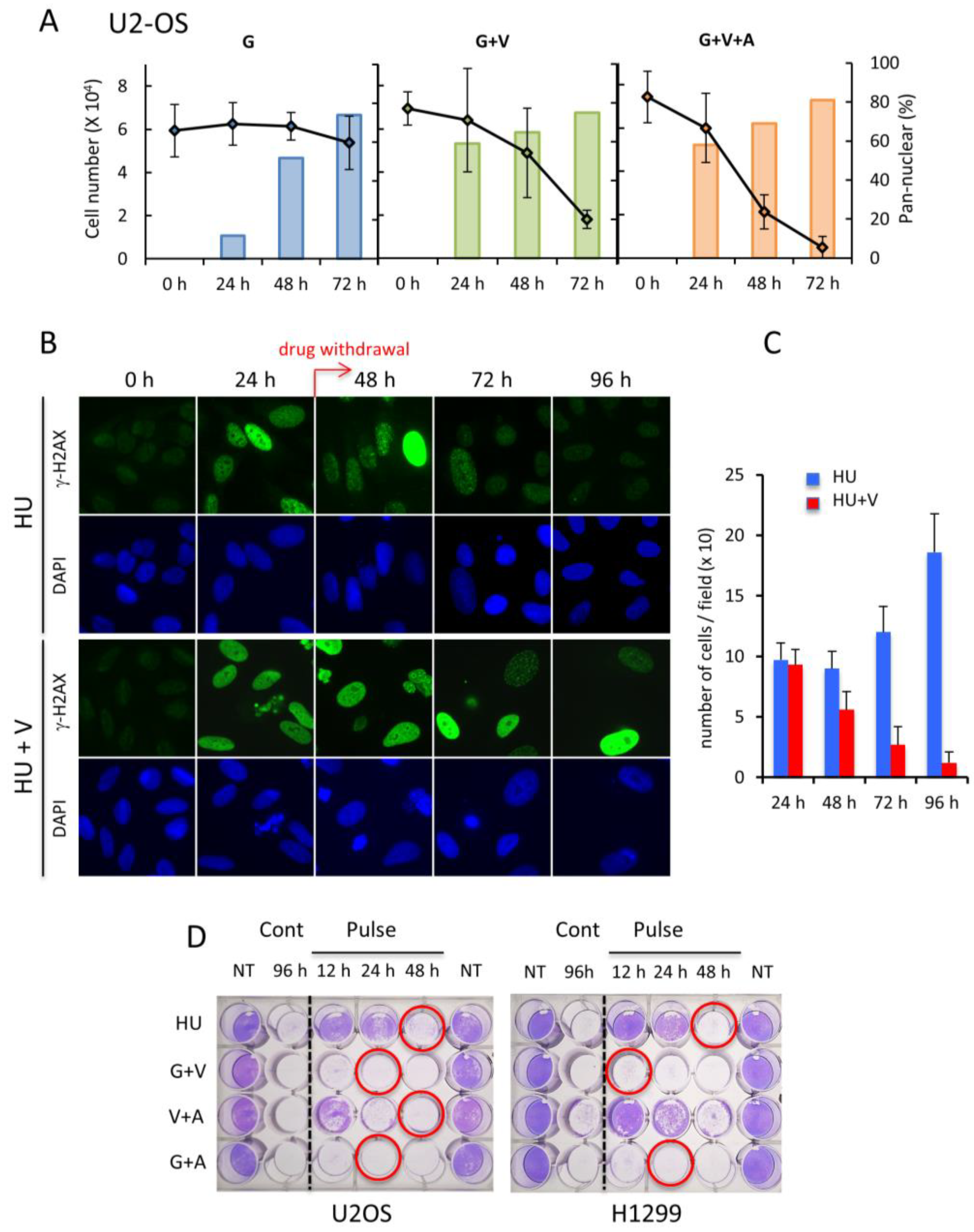
2.4. Pan-Nuclear γ-H2AX Staining Is Correlated with the Loss of Cell Viability
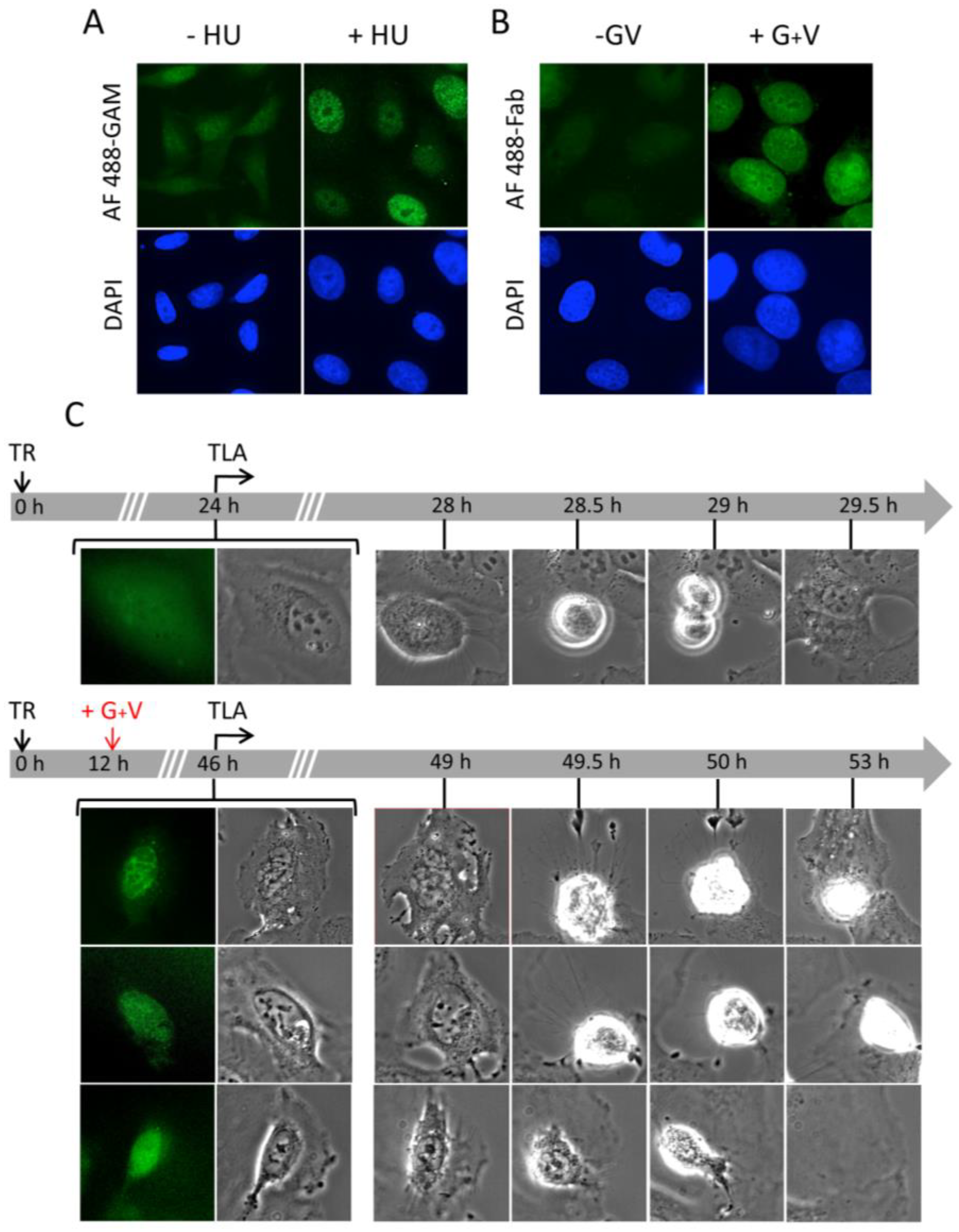
2.5. Pan-Nuclear γ-H2AX Pattern Is a Signature of Induced Cell Death and Thus of Lethal Replication Stress
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Culture and Assays
4.3. Generation of the H2AFX−/− Cell Lines
4.4. Western Blot and ELISA
4.5. Immunofluorescence Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Redon, C.E.; Weyemi, U.; Parekh, P.R.; Huang, D.; Burrell, A.S.; Bonner, W.M. γ-H2AX and other histone post-translational modifications in the clinic. Biochim. Biophys. Acta 2012, 1819, 743–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Ward, I.M.; Chen, J. Histone H2AX Is Phosphorylated in an ATR-dependent Manner in Response to Replicational Stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of Dynamic γ-H2AX Domains along Broken DNA Strands Is Distinctly Regulated by ATM and MDC1 and Dependent upon H2AX Densities in Chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef]
- Nikolova, T.; Dvorak, M.; Jung, F.; Adam, I.; Krämer, E.; Gerhold-Ay, A.; Kaina, B. The γH2AX Assay for Genotoxic and Nongenotoxic Agents: Comparison of H2AX Phosphorylation with Cell Death Response. Toxicol. Sci. 2014, 140, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef]
- Buisson, I.; Le Bouffant, R.; Futel, M.; Riou, J.-F.; Umbhauer, M. Pax8 and Pax2 are specifically required at different steps of Xenopus pronephros development. Dev. Biol. 2015, 397, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Sarni, D.; Kerem, B. Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis. Int. J. Mol. Sci. 2017, 18, 1339. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 17, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, Y.; Wang, J.; Zhang, X.; Gao, Y.; Yin, L.; Li, Q.; Li, J.; Chen, H. Induction and inhibition of the pan-nuclear gamma-H2AX response in resting human peripheral blood lymphocytes after X-ray irradiation. Cell Death Discov. 2016, 2, 16011. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Parsels, J.D.; Tanska, D.M.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. The contribution of DNA replication stress marked by high-intensity, pan-nuclear γH2AX staining to chemosensitization by CHK1 and WEE1 inhibitors. Cell Cycle 2018, 17, 1076–1086. [Google Scholar] [CrossRef]
- Quanz, M.; Chassoux, D.; Berthault, N.; Agrario, C.; Sun, J.-S.; Dutreix, M. Hyperactivation of DNA-PK by Double-Strand Break Mimicking Molecules Disorganizes DNA Damage Response. PLoS ONE 2009, 4, e6298. [Google Scholar] [CrossRef]
- Meyer, B.; Voss, K.-O.; Tobias, F.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Clustered DNA damage induces pan-nuclear H2AX phosphorylation mediated by ATM and DNA–PK. Nucleic Acids Res. 2013, 41, 6109–6118. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Brady, D.; Prise, K. Alpha particles induce pan-nuclear phosphorylation of H2AX in primary human lymphocytes mediated through ATM. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 2199–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desplancq, D.; Freund, G.; Conic, S.; Sibler, A.-P.; Didier, P.; Stoessel, A.; Oulad-Abdelghani, M.; Vigneron, M.; Wagner, J.; Mély, Y.; et al. Targeting the replisome with transduced monoclonal antibodies triggers lethal DNA replication stress in cancer cells. Exp. Cell Res. 2016, 342, 145–158. [Google Scholar] [CrossRef]
- Singh, A.; Xu, Y.-J. The Cell Killing Mechanisms of Hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef]
- Freund, G.; Sibler, A.-P.; Desplancq, D.; Oulad-Abdelghani, M.; Vigneron, M.; Gannon, J.; Van Regenmortel, M.H.; Weiss, E. Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. mAbs 2013, 5, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Conic, S.; Desplancq, D.; Tora, L.; Weiss, E. Electroporation of Labeled Antibodies to Visualize Endogenous Proteins and Posttranslational Modifications in Living Metazoan Cell Types. Bio Protoc. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Ewald, B.; Sampath, D.; Plunkett, W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol. Cancer Ther. 2007, 6, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.-Y.; Obuse, C.; Nishi, R.; et al. BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef]
- Grimaudo, S.; Tolomeo, M.; Chimirri, A.; Zappala, M.; Gancitano, R.A.; D’Alessandro, N. Selective induction of apoptosis in multidrug resistant HL60R cells by the thiazolobenzoimidazole derivative 1-(2,6-difluorophenyl)-1H,3H-thiazolo [3,4-a] benzimidazole (TBZ). Eur. J. Cancer 1998, 34, 1756–1763. [Google Scholar] [CrossRef]
- Fernandez-Vidal, A.; Vignard, J.; Mirey, G. Around and beyond 53BP1 Nuclear Bodies. Int. J. Mol. Sci. 2017, 18, 2611. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Ashley, A.K.; Shrivastav, M.; Nie, J.; Amerin, C.; Troksa, K.; Glanzer, J.G.; Liu, S.; Opiyo, S.O.; Dimitrova, D.D.; Le, P.; et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair 2014, 21, 131–139. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Conic, S.; Desplancq, D.; Ferrand, A.; Fischer, V.; Heyer, V.; San Martin, B.R.; Pontabry, J.; Oulad-Abdelghani, M.; Wright, G.D.; Molina, N.; et al. Imaging of native transcription factors and histone phosphorylation at high resolution in live cells. J. Cell Biol. 2018, 217, 1537–1552. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Zhang, Y.; Redon, C.E.; Reinhold, W.C.; Chen, A.P.; Fogli, L.K.; Holbeck, S.L.; Parchment, R.E.; Hollingshead, M.; Tomaszewski, J.E.; et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS ONE 2017, 12, e0171582. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Muslimovic, A.; Hultborn, R.; Fernström, E.; Hammarsten, O. In-solution staining and arraying method for the immunofluorescence detection of γH2AX foci optimized for clinical applications. BioTechniques 2011, 51, 185–189. [Google Scholar] [CrossRef]
- Fu, S.; Yang, Y.; Tirtha, D.; Yen, Y.; Zhou, B.; Zhou, M.-M.; Ohlmeyer, M.; Ko, E.C.; Cagan, R.; Rosenstein, B.S.; et al. γ-H2AX Kinetics as a Novel Approach to High Content Screening for Small Molecule Radiosensitizers. PLoS ONE 2012, 7, e38465. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the γ-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef]
- Liaw, H.; Lee, D.; Myung, K. DNA-PK-Dependent RPA2 Hyperphosphorylation Facilitates DNA Repair and Suppresses Sister Chromatid Exchange. PLoS ONE 2011, 6, e21424. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Kessinger, C.; Kobayashi, J.; Chen, B.P.C.; Chen, D.J.; Chatterjee, A.; Burma, S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 2006, 5, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Zuazua-Villar, P.; Ganesh, A.; Phear, G.; Gagou, M.E.; Meuth, M. Extensive RPA2 hyperphosphorylation promotes apoptosis in response to DNA replication stress in CHK1 inhibited cells. Nucleic Acids Res. 2015, 43, 9776–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, L.-A.; Kumar, A.; Stirling, P. Chromatin as a Platform for Modulating the Replication Stress Response. Genes 2018, 9, 622. [Google Scholar] [CrossRef] [PubMed]
- Revet, I.; Feeney, L.; Bruguera, S.; Wilson, W.; Dong, T.K.; Oh, D.H.; Dankort, D.; Cleaver, J.E. Functional relevance of the histone H2Ax in the response to DNA damaging agents. Proc. Natl. Acad. Sci. USA 2011, 108, 8663–8667. [Google Scholar] [CrossRef] [Green Version]
- Gagou, M.E.; Zuazua-Villar, P.; Meuth, M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell 2010, 21, 739–752. [Google Scholar] [CrossRef]
- Goss, K.L.; Koppenhafer, S.L.; Harmoney, K.M.; Terry, W.W.; Gordon, D.J. Inhibition of CHK1 sensitizes Ewing sarcoma cells to the ribonucleotide reductase inhibitor gemcitabine. Oncotarget 2017, 8, 87016–87032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imreh, G.; Norberg, H.V.; Imreh, S.; Zhivotovsky, B. Chromosomal breaks during mitotic catastrophe trigger γH2AX-ATM-p53-mediated apoptosis. J. Cell Sci. 2016, 129, 1950. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-B.; Courtin, A.; Boyce, R.J.; Boyle, R.G.; Richards, F.M.; Jodrell, D.I. CHK1 Inhibition Synergizes with Gemcitabine Initially by Destabilizing the DNA Replication Apparatus. Cancer Res. 2015, 75, 3583–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef]
- De Feraudy, S.; Revet, I.; Bezrookove, V.; Feeney, L.; Cleaver, J.E. A minority of foci or pan-nuclear apoptotic staining of H2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Plappert-Helbig, U.; Libertini, S.; Frieauff, W.; Theil, D.; Martus, H.-J. Gamma-H2AX immunofluorescence for the detection of tissue-specific genotoxicity in vivo: Gamma-H2AX Tissue-specific Genotoxicity. Environ. Mol. Mutagen. 2019, 60, 4–16. [Google Scholar] [CrossRef]
- Andrew, S.M.; Titus, J.A. Fragmentation of immunoglobulin G. Curr. Protoc. Cell Biol. 2003, 17, 16-4. [Google Scholar] [CrossRef]
- Dietsch, F.; Donzeau, M.; Cordonnier, A.M.; Weiss, E.; Chatton, B.; Vigneron, M. A fast method for analyzing essential protein mutants in human cells. BioTechniques 2017, 62, 80–82. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moeglin, E.; Desplancq, D.; Conic, S.; Oulad-Abdelghani, M.; Stoessel, A.; Chiper, M.; Vigneron, M.; Didier, P.; Tora, L.; Weiss, E. Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress. Cancers 2019, 11, 355. https://doi.org/10.3390/cancers11030355
Moeglin E, Desplancq D, Conic S, Oulad-Abdelghani M, Stoessel A, Chiper M, Vigneron M, Didier P, Tora L, Weiss E. Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress. Cancers. 2019; 11(3):355. https://doi.org/10.3390/cancers11030355
Chicago/Turabian StyleMoeglin, Eric, Dominique Desplancq, Sascha Conic, Mustapha Oulad-Abdelghani, Audrey Stoessel, Manuela Chiper, Marc Vigneron, Pascal Didier, Laszlo Tora, and Etienne Weiss. 2019. "Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress" Cancers 11, no. 3: 355. https://doi.org/10.3390/cancers11030355
APA StyleMoeglin, E., Desplancq, D., Conic, S., Oulad-Abdelghani, M., Stoessel, A., Chiper, M., Vigneron, M., Didier, P., Tora, L., & Weiss, E. (2019). Uniform Widespread Nuclear Phosphorylation of Histone H2AX Is an Indicator of Lethal DNA Replication Stress. Cancers, 11(3), 355. https://doi.org/10.3390/cancers11030355