Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation



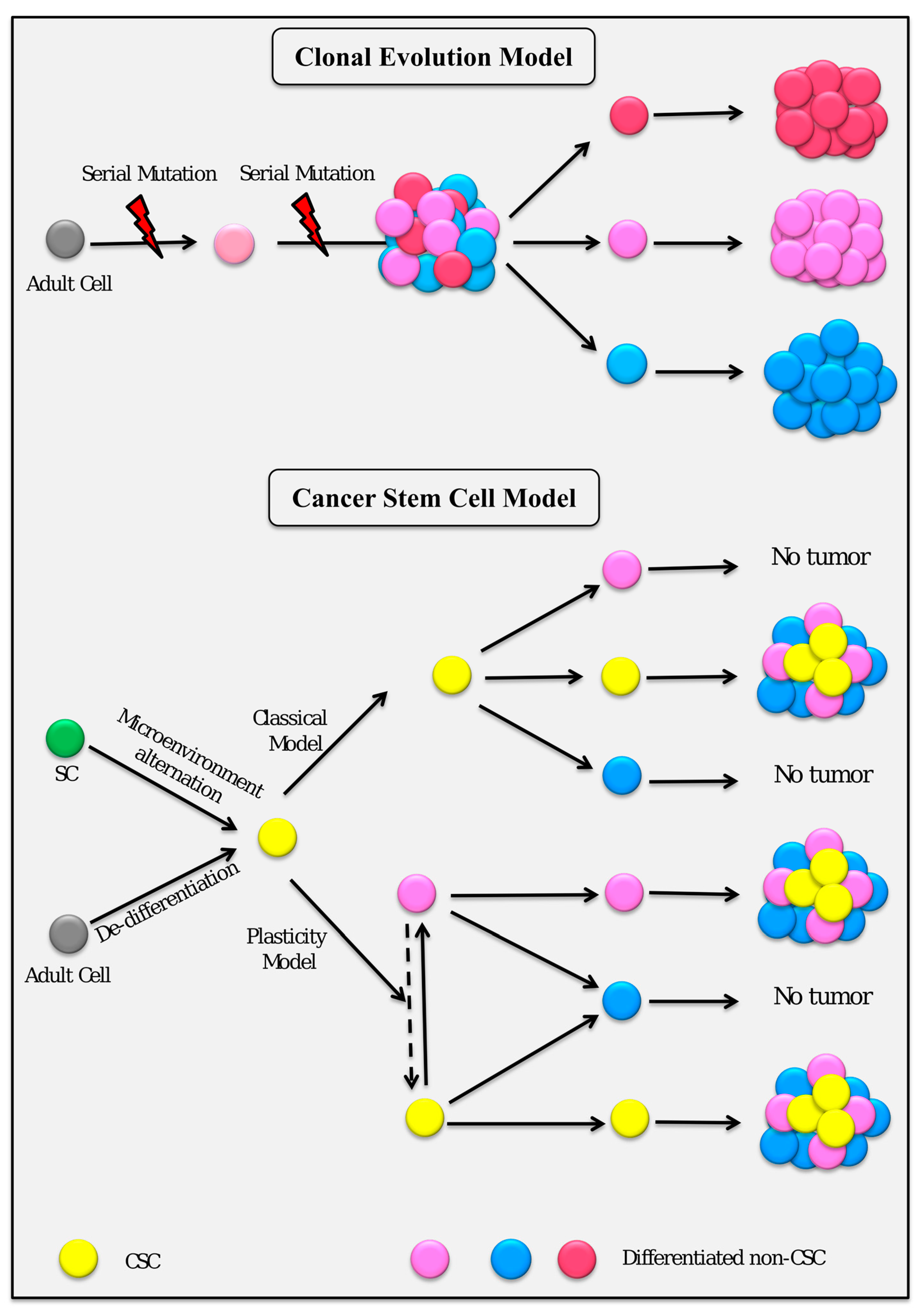{kind=link}
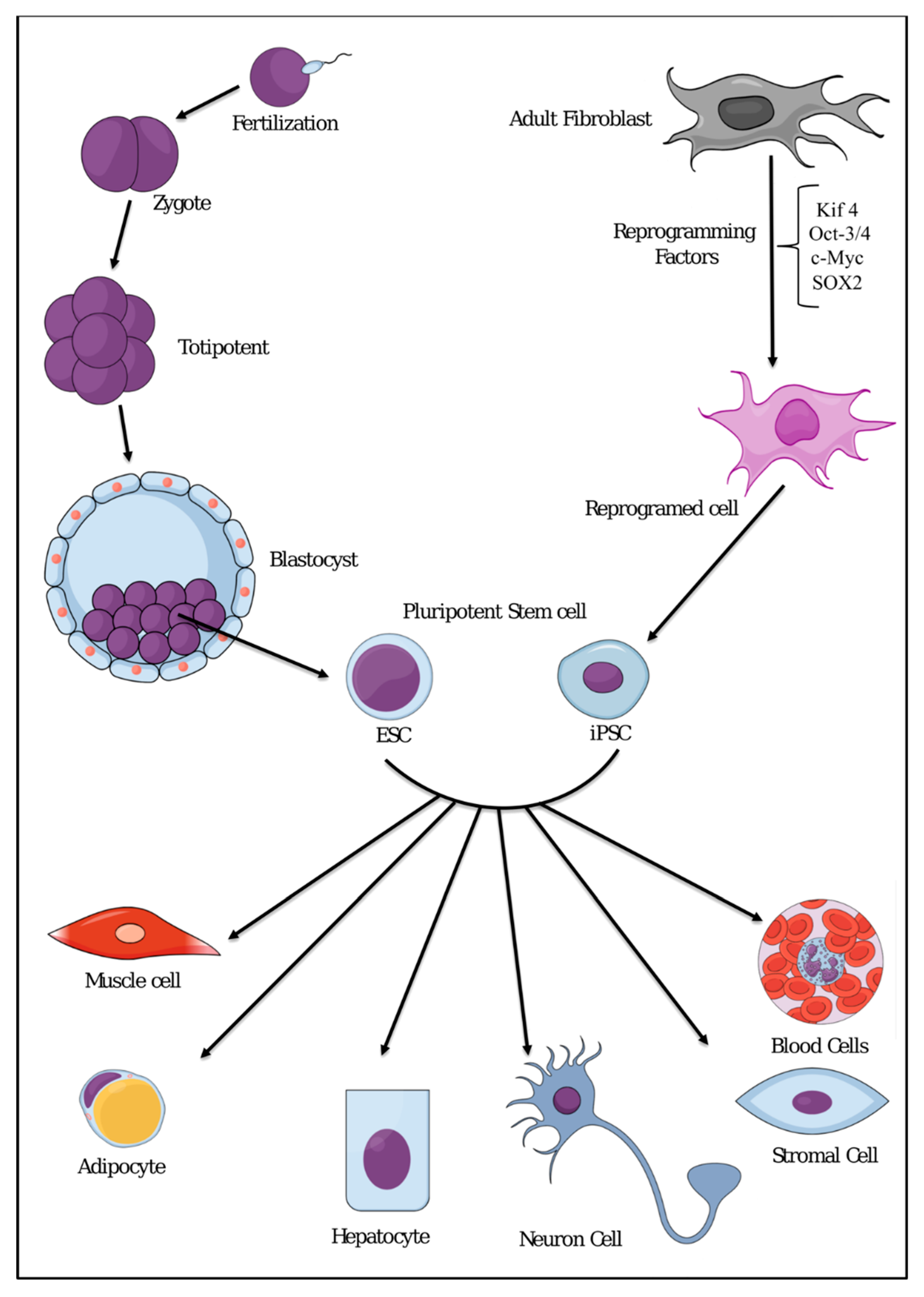{kind=link}
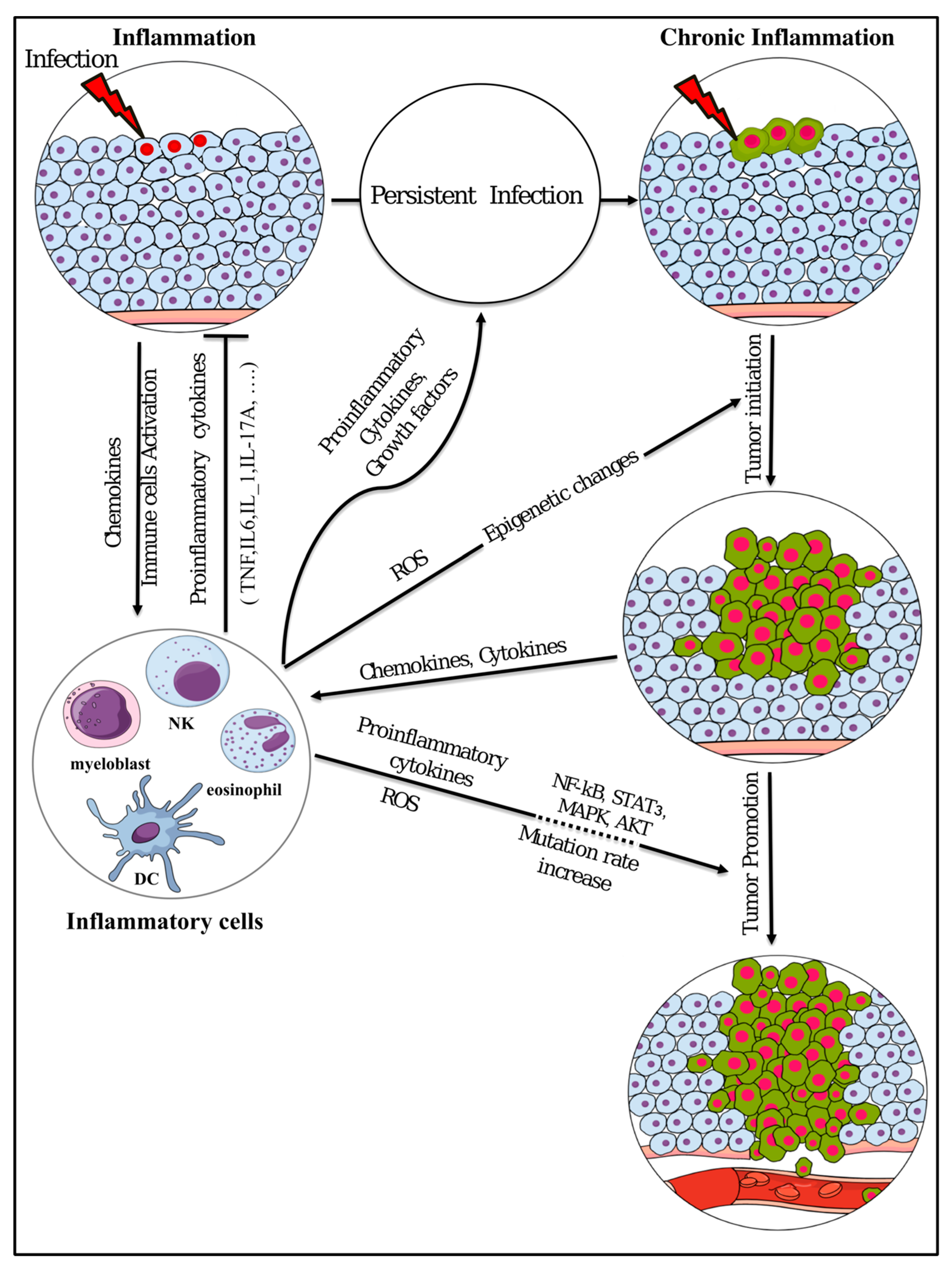{kind=link}
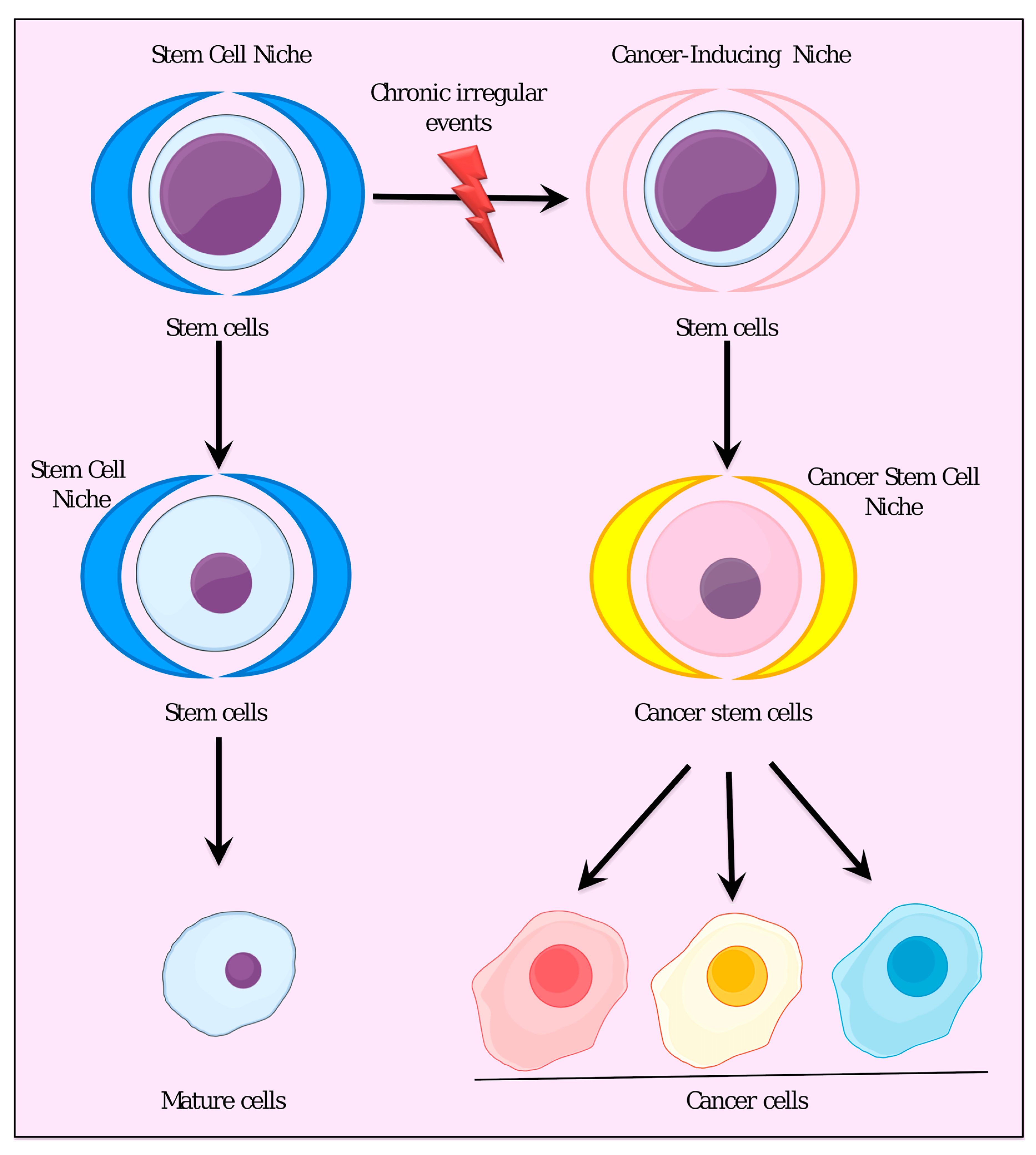{kind=link}
{kind=link}
Abstract
1. Introduction
2. Inflammatory Microenvironment Stimulation and Cancer
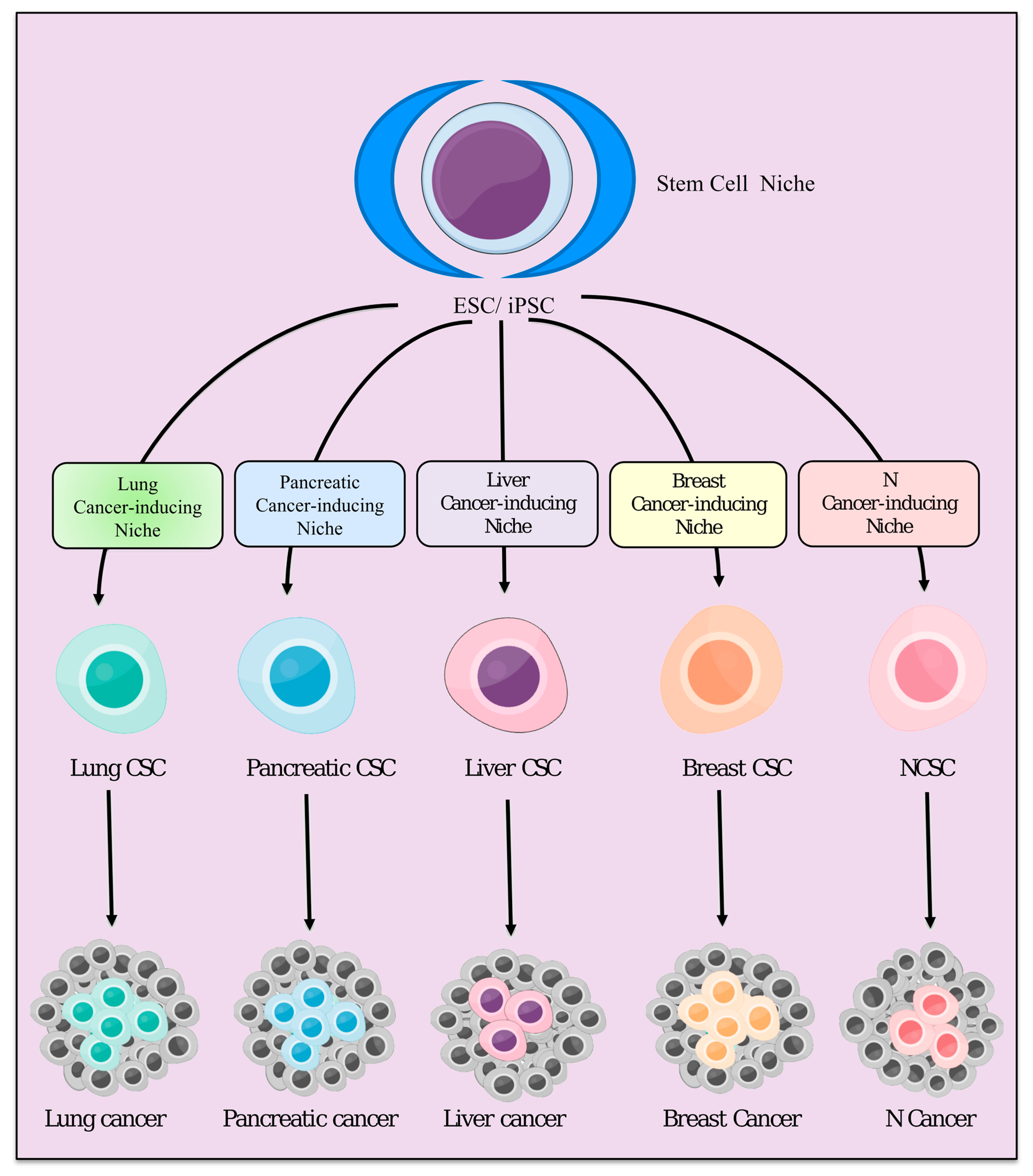
3. Stem Cell Niche and Cancer-Inducing Niche
4. Inflammatory Microenvironment Stimulates the Generation of Cancer Stem Cells
4.1. Lung Cancer Stem Cells
4.2. Pancreatic Cancer Stem Cells
4.3. Liver Cancer Stem Cells
4.4. Prostate Cancer Stem Cells
5. Epigenesis or Mutagenesis?
5.1. Hyper- and Hypo- Methylation of DNA
5.2. DNA Mutations
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| CSCs | Cancer stem cells |
| iPSCs | Induced pluripotent stem cells |
| CICs | Cancer-initiating cells |
| ROS | Reactive oxygen species |
| DCs | Dendritic cells |
| ESC | Embryonic stem cell |
| MSCs | Mesenchymal stem cells |
| CM | Conditioned medium |
| PDAC | Pancreatic duct adenocarcinoma |
| ROS | Reactive oxygen species |
| SCLC | Small cell lung cancer |
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L. Genome Remodeling in a Basal-like Breast Cancer Metastasis and Xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Cho, R.J.; Gray, J.W. What are we learning from the cancer genome? Nat. Rev. Clin. Oncol. 2012, 9, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Zur Frage der Entstehung maligner Tumoren; Verlag von Gustav Fischer: Jena, Germany, 1914; pp. 29–32. [Google Scholar]
- Cobb, M. When genes become “information”. Cell 2013, 153, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Nordling, C.O. A new theory on the cancer-inducing mechanism. Br. J. Cancer 1953, 7, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.J.B. The two “hit” and multiple “hit” theories of carcinogenesis. Br. J. Cancer 1969, 23, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004, 6, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L. In defense of the somatic mutation theory of cancer. Bioessays 2011, 3, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Novelli, M.R.; Bodmer, W.F. The mutation rate and cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 14800–14803. [Google Scholar] [CrossRef]
- Versteeg, R. Tumours outside the mutation box. Nature 2014, 506, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Sherley, J.L. Asymmetric cell kinetics genes: The key to expansion of adult stem cells in culture. Stem Cells 2002, 20, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Inaba, M.; Yamashita, Y.M. Asymmetric stem cell division: Precision for robustness. Cell Stem Cell 2012, 11, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Perotti, C.; Del Fante, C.; Cervio, M.; Redi, C.A. Fondazione IRCCS Policlinico San Matteo, Pavia (Italia). Stem cells: Sources and therapies. Biol. Res. 2012, 45, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Maximow, A. Der Lymphozyt als gemeinsame Stammzelle der verschiedenen Blutelemente in der embryonalen Entwicklung und im postfetalen Leben der Säugetiere (Demonstrationsvortrag, gehalten in der außerordentlichen Sitzung der Berliner Hämatologischen Gesellschaft am 1. Juni 1909). Folia Haematol. 2009, 125–134. [Google Scholar]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [PubMed]
- Cregan, M.D.; Fan, Y.; Appelbee, A.J.; Brown, M.L.; Klopcic, B.; Koppen, J.A.; Mitoulas, L.R.; Piper, K.M.E.; Choolani, M.A.; Chong, Y.S.; et al. Identification of nestin-positive putative mammary stem cells in human breast milk. Cell Tissue Res. 2007, 329, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Kaufman, M. Establishment in culture of pluripotent cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Mountford, J. Human embryonic stem cells: Origins, characteristics and potential for regenerative therapy. Transfus. Med. 2008, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Heath, J.K.; Donaldson, D.D.; Wong, G.G.; Moreau, J.; Stahl, M.; Rogers, D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 1988, 336, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Cavaleri, F.; Scholer, H.R. Nanog: A new recruit to the embryonic stem cell orchestra. Cell 2003, 113, 551–552. [Google Scholar] [CrossRef]
- Patil, A.M. Embryonic Stem Cell Research Ethical and Legal Controversies. J. Indian Acad. Forensic Med. 2014, 36, 188–194. [Google Scholar]
- Brücher, B.L.; Jamall, I.S. Somatic Mutation Theory—Why it’s Wrong for Most Cancers. Cell Physiol. Biochem. 2016, 38, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Somatic mutation theory of carcinogenesis: Why it should be dropped and replaced. Mol. Carcinogens. 2000, 29, 205–211. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, S.C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Li, W.; Lv, Z.; Liu, L.; Tong, M.; Hai, T.; Hao, J.; Guo, C.L.; Ma, Q.W.; Wang, L.; et al. iPS cells produce viable mice through tetraploid complementation. Nature 2009, 461, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Wang, J.; Zhang, Y.; Kou, Z.; Gao, S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell 2009, 5, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Hazen, J.L.; Nazor, K.L.; Rodriguez, A.R.; Gifford, W.; Martin, G.; Kupriyanov, S.; Baldwin, K.K. Adult mice generated from induced pluripotent stem cells. Nature 2009, 461, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Hochedlinger, K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat. Cell Biol. 2011, 13, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die krankhaften Geschwülste. Dreissig Vorlesungen, gehalten wahrend des Wintersemesters 1862–1863 an Der Universität Zu Berlin; Hirschwald: Berlin, Germany, 1863; p. 69. [Google Scholar]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Li, J.D.Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 23, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Dassoler, M.; Schwanz, M.; Busseto, F.; Moreira, E.A.; Gutierrez, L. Perfil fitoquímico e ensaio farmacológico de Averrhoa carambola L. (Oxalidaceae). J. Bras. Fitom. 2004, 2, 4–8. [Google Scholar]
- Falcão, H.; Lima, I.O.; Santos, V.L.; Dantas, H.F.; Diniz, M.F.F.M.; Barbosa-Filho, J.M.; Batista, L.M. Review of the plants with anti-inflammatory activity studied in Brazil. Braz. J. Pharmacogn. 2005, 15, 381–391. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Greten, FR.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKb links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaihan, D. Epigenetic Mechanisms in Inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313 (Pt 1), 17–29. [Google Scholar] [CrossRef]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxid. Med. Cell. 2013, 387014. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xie, T. Stem Cell Niche: Structure and Function. Annu. Rev. Cell Dev. Biol. 2005, 21, 605–631. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Lemischka, I.R. Stem Cells and Their Niches. Science 2006, 311, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Scadden, D.T. Nice neighborhood: Emerging concepts of the stem cell niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Sato, T.; van de Wetering, M.; Lyubimova, A.; Yee Nee, A.N.; Gregorieff, A.; Sasaki, N.; Zeinstra, L.; van den Born, M.; Korving, J.; et al. Dll1+secretory progenitor cell srevertto stem cells upon cryptdamage. Nat. Cell Biol. 2012, 14, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Jones, D.L.; Wagers, A.J. No place like home: Anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 2008, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sells, S. On the Stem Cell Origin of Cancer. Am. J. Pathol. 2010, 176, 2584–2594. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, F.D. Mutations differ in normal and cancer cells of the oesophagus. Nature 2019, 565, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic Stem Cells: Sources, Niches, and Vital Pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumor Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Yan, T.; Mizutani, A.; Sota, T.; Hiramoto, Y.; Prieto-Vila, M.; Chen, L.; Satoh, A.; Kudoh, T.; Kasai, T.; et al. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int. J. Cancer 2014, 135, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Anna Sanchez Calle, A.S.; Zahra, M.H.; Prieto-Vila, M.; Oo, A.K.K.; Hurley, L.; Vaidyanath, A.; Seno, A.; Masuda, J.; Iwasaki, Y.; et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci. Rep. 2017, 7, 6838. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The cancer stem cell niche—There goes the neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Calle, A.S.; Nair, N.; Oo, A.K.; Prieto-Vila, M.; Koga, M.; Khayrani, A.C.; Hussein, M.; Hurley, L.; Vaidyanath, A.; Seno, A.; et al. A new PDAC mouse model originated from iPSCs-converted pancreatic cancer stem cells (CSCcm). Am. J. Cancer Res. 2016, 6, 2799–2815. [Google Scholar] [PubMed]
- Afify, S.M.; Calle, A.S.; Kumon, K.; Nawara, H.M.; Khairani, A.C.; Mahmud, H.; Oo, A.K.K.; Juan, D.; Zahara, M.H.; Seno, A.; et al. A model of CSC converted from iPSC in the conditioned medium of HCC paving the way to establish HCC CSC [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS ONE 2012, 7, e33544. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.B.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.N.; Gazdar, A.F.; Minna, J.D. Positive correlation between histological tumor involvement and generation of tumor cell colonies in agarose in specimens taken directly from patients with small cell carcinoma of the lung. Cancer Res. 1980, 40, 1820–1823. [Google Scholar] [PubMed]
- Carney, D.N.; Gazdar, A.F.; Bunn, P.A. Demonstration of the stem cell nature of clonogenic tumor cells from lung cancer patients. Stem Cells 1982, 1, 149–164. [Google Scholar] [PubMed]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Mizutani, A.; Chen, L.; Takaki, M.; Hiramoto, Y.; Matsuda, S.; Shigehiro, T.; Kasai, T.; Kudoh, T.; Murakami, H.; et al. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J. Cancer 2014, 5, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Oo, A.K.K.; Calle, A.S.; Nair, N.; Mahmud, H.; Vaidyanath, A.; Yamauchi, J.; Khayrani, A.C.; Du, J.; Alam, M.J.; Seno, A.; et al. Up-Regulation of PI 3-Kinases and the Activation of PI3K-Akt Signaling Pathway in Cancer Stem-Like Cells Through DNA Hypomethylation Mediated by the Cancer Microenvironment. Transl. Oncol. 2018, 11, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Valle, S.; Martin-Hijano, L.; Alcalá, S.; Alonso-Nocelo, M.; Sainz, J. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, S.; Alcala, S.; Fernandez, G.; Bou Kheir, T.; Schoenhals, M.; González-Neira, A.; Fernandez, F.M.; Aicher, A.; Heeschen, C.; et al. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17-92 Cluster. Cancer Res. 2011, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Alcala, S.; Usachov, V.; Hermann, P.C.; Sainz, B., Jr. The ever-changing landscape of pancreatic cancer stem cells. Pancreatology 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Kassebaum, N.J.; Bertozzi-Villa, A.; Coggeshall, M.S.; Shackelford, K.A.; Steiner, C.; Heuton, K.R. Global, regional, and national levels and causes of maternal mortality during 1990–2013: A systematic analysis for the Global Burden of Disease Study. Lancet 2014, 384, 980–1004. [Google Scholar] [CrossRef]
- Chiba, T.; Zheng, Y.W.; Kita, K.; Yokosuka, O.; Saisho, H.; Onodera, M.; Miyoshi, H.; Nakano, M.; Zen, Y.; Nakanuma, Y.; et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 2007, 133, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Z.; Yu, X.H. Bone marrow cells: The source of hepatocellular carcinoma? Med. Hypotheses. 2007, 69, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, C.; Han, K.H.; Choi, J.; Kim, Y.B.; Kim, J.K.; Park, Y.N. Primary liver carcinoma of intermediate (hepatocyte-cholangiocyte) phenotype. J. Hepatol. 2004, 40, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, X.P.; Zhang, W.; Dong, H.H.; Xiang, S.; Zhang, W.G.; Zhang, B.X. Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: Immunohistochemical and double-fluorescence immunostaining evidence. Histopathology 2008, 52, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Invest. 2013, 123, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; Yasuda, K.; Fukumitsu, K.; Mizumoto, M.; et al. Keratin 19, a Cancer Stem Cell Marker in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2015, 21, 3081–3091. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Fu, D.; Ma, Y.; Shen, X.Z. The power and the promise of liver cancer stem cell markers. Stem Cells Dev. 2011, 20, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Hernandez, J.C.; Dean, A.M.; Rao, P.H.; Darlington, G.J. CD24-positive cells from normal adult mouse liver are hepatocyte progenitor cells. Stem Cells Dev. 2011, 20, 2177–2188. [Google Scholar] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Huss, W.J.; Gray, D.R.; Greenberg, N.M. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005, 65, 6640–6650. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Good cells gone bad: The cellular origins of cancer. Trends Mol. Med. 2010, 16, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, X.; Watanabe, M.; Ueki, H.; Hu, H.; Li, N.; Araki, M.; Wada, K.; Xu, A.; Liu, C.; et al. Induction of cells with prostate cancer stem-like properties from mouse induced pluripotent stem cells via conditioned medium. Am. J. Cancer Res. 2018, 8, 1624–1632. [Google Scholar] [PubMed]
- Seno, A.; Kasai, T.; Ikeda, M.; Vaidyanath, A.; Masuda, J.; Mizutani, A.; Murakami, H.; Ishikawa, T.; Seno, M. Characterization of Gene Expression Patterns among Artificially Developed Cancer Stem Cells Using Spherical Self-Organizing Map. Cancer Inform. 2016, 15, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. Epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Vaissiere, T.; Herceg, Z. Histone acetylation and chromatin signature in stem cell identity and cancer. Mutat. Res. 2008, 637, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Delpu, Y.; Cordelier, P.; Cho, W.; Torrisani, J. DNA Methylation and Cancer Diagnosis. Int. J. Mol. Sci. 2013, 14, 15029–15058. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Wild, L.; Flanagan, J.M. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim. Biophys. Acta 2010, 1806, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dai, D.; Chen, B.; Tang, H.; Xie, X.; Wei, W. Efficacy of PI3K/AKT/mTOR pathway inhibitors for the treatment of advanced solid cancers: A literature-based meta-analysis of 46 randomised control trials. PLoS ONE 2018, 13, e0192464. [Google Scholar] [CrossRef] [PubMed]
- Jacqueline, C.; Biro, P.A.; Beckmann, C.; Moller, A.P.; Renaud, F.; Sorci, G.; Tasiemski, A.; Ujvari, B.; Thomas, F. Cancer: A disease at the crossroads of trade-offs. Evol. Appl. 2016, 10, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Adjiri, A. DNA Mutations May Not Be the Cause of Cancer. Oncol. Ther. 2017, 5, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Adjiri, A. Identifying and targeting the cause of cancer is needed to cure cancer. Oncol. Ther. 2016, 4, 17–33. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afify, S.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. https://doi.org/10.3390/cancers11030345
Afify SM, Seno M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers. 2019; 11(3):345. https://doi.org/10.3390/cancers11030345
Chicago/Turabian StyleAfify, Said M., and Masaharu Seno. 2019. "Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation" Cancers 11, no. 3: 345. https://doi.org/10.3390/cancers11030345
APA StyleAfify, S. M., & Seno, M. (2019). Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers, 11(3), 345. https://doi.org/10.3390/cancers11030345