Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L As A Novel Anti-Metastatic Enzyme Cancer Therapy


 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
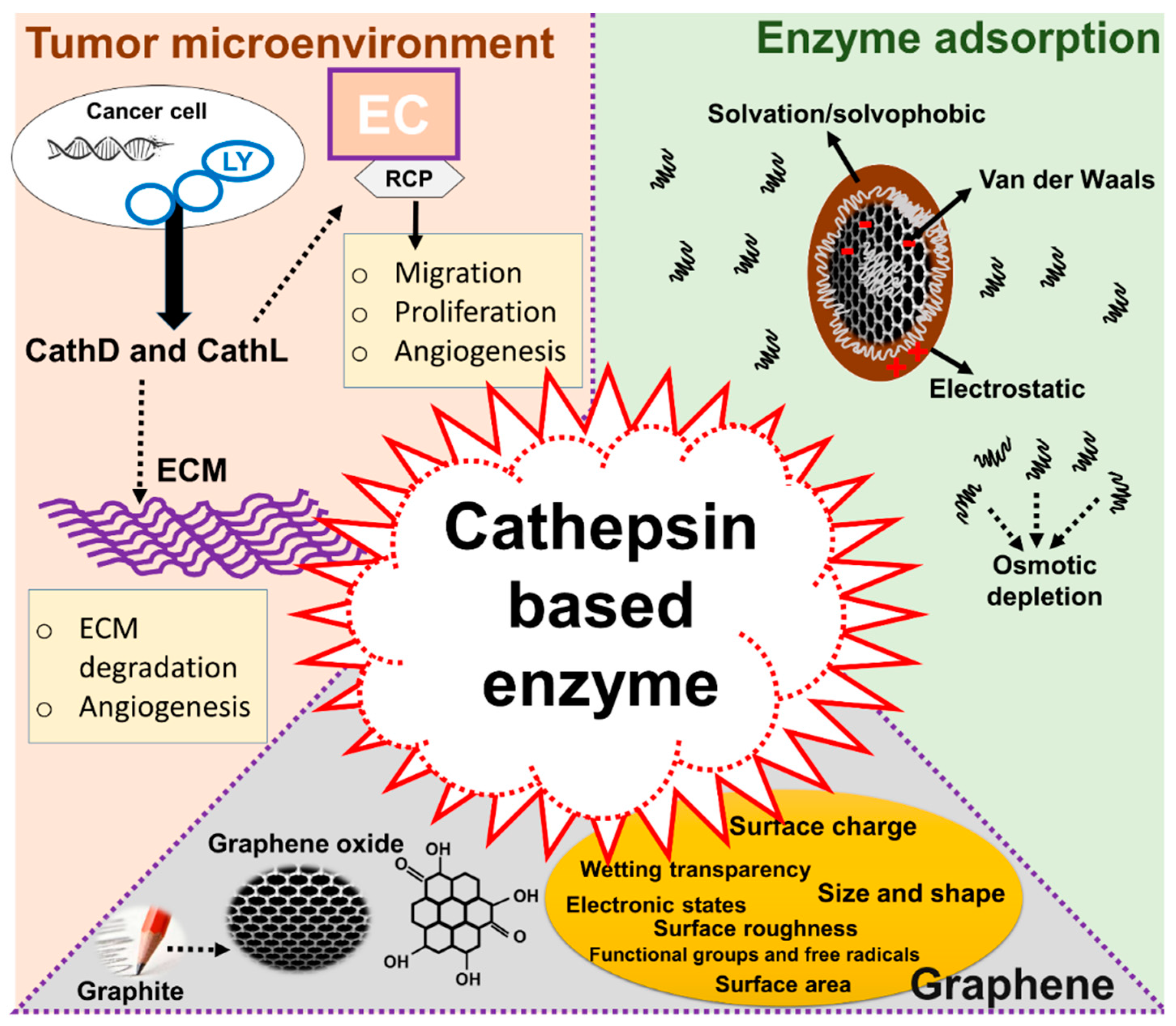
1. Introduction
2. Results
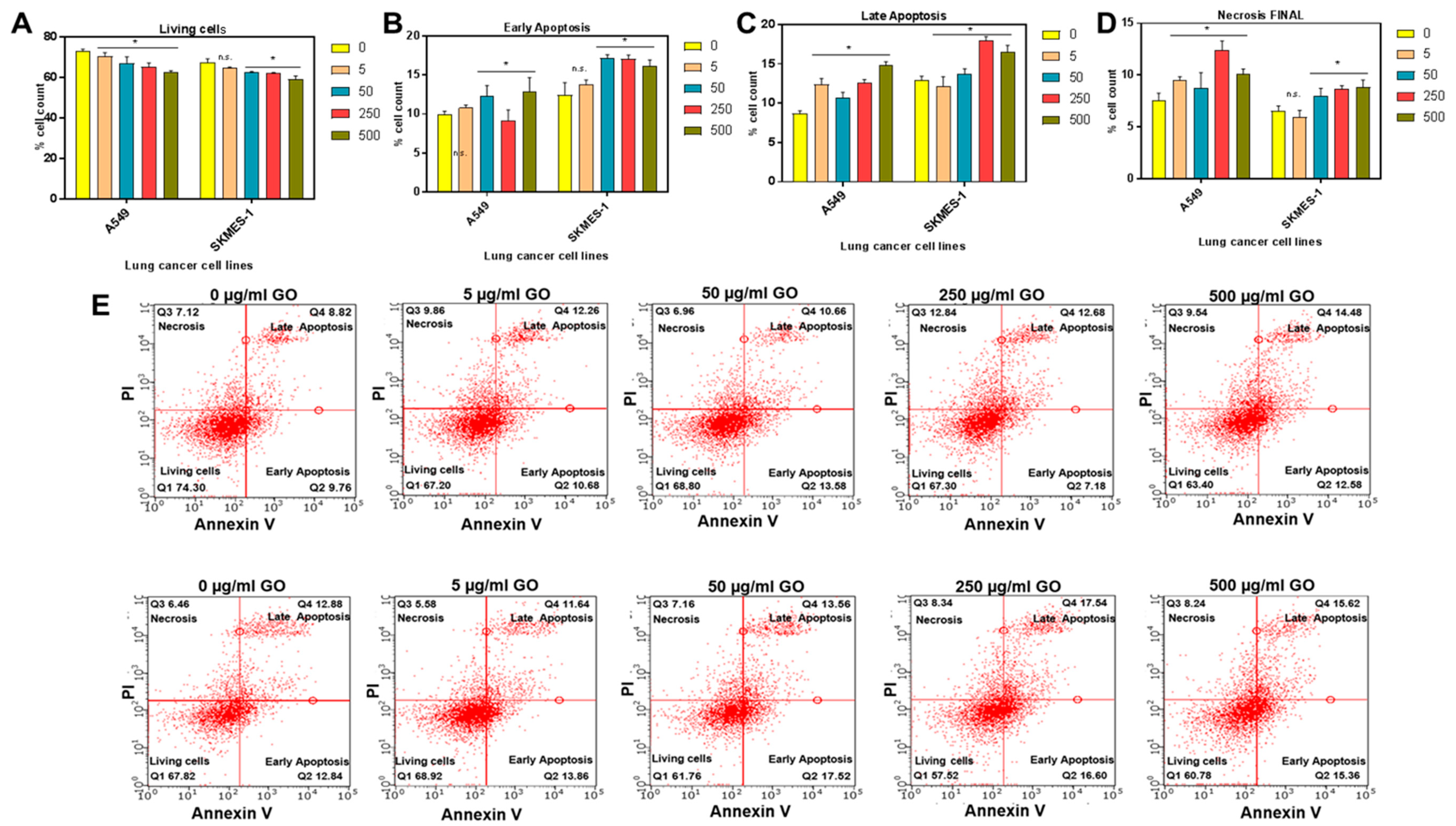
2.1. Synthesis and In Vitro Toxic Effects of GO on Lung Cancer Cells
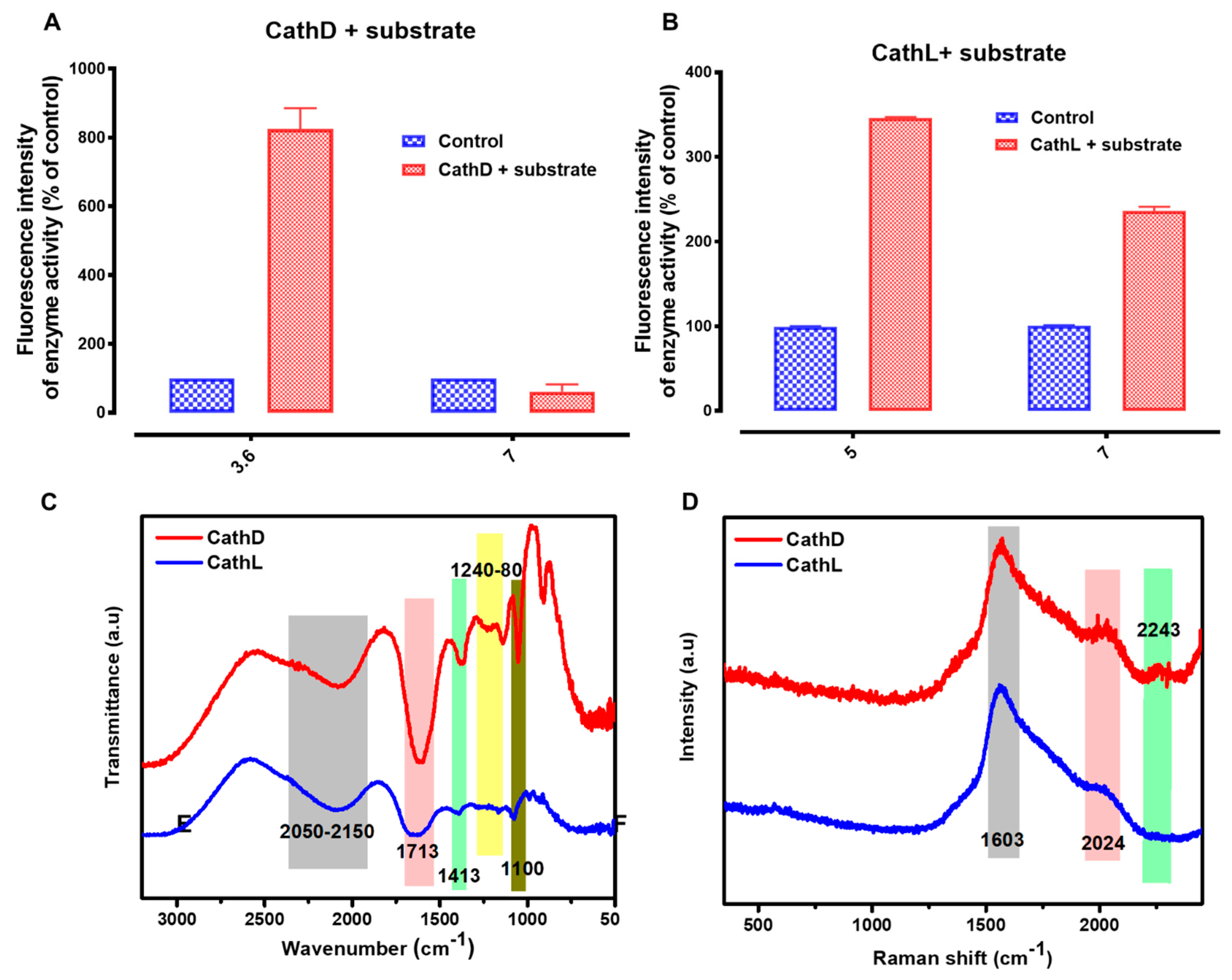
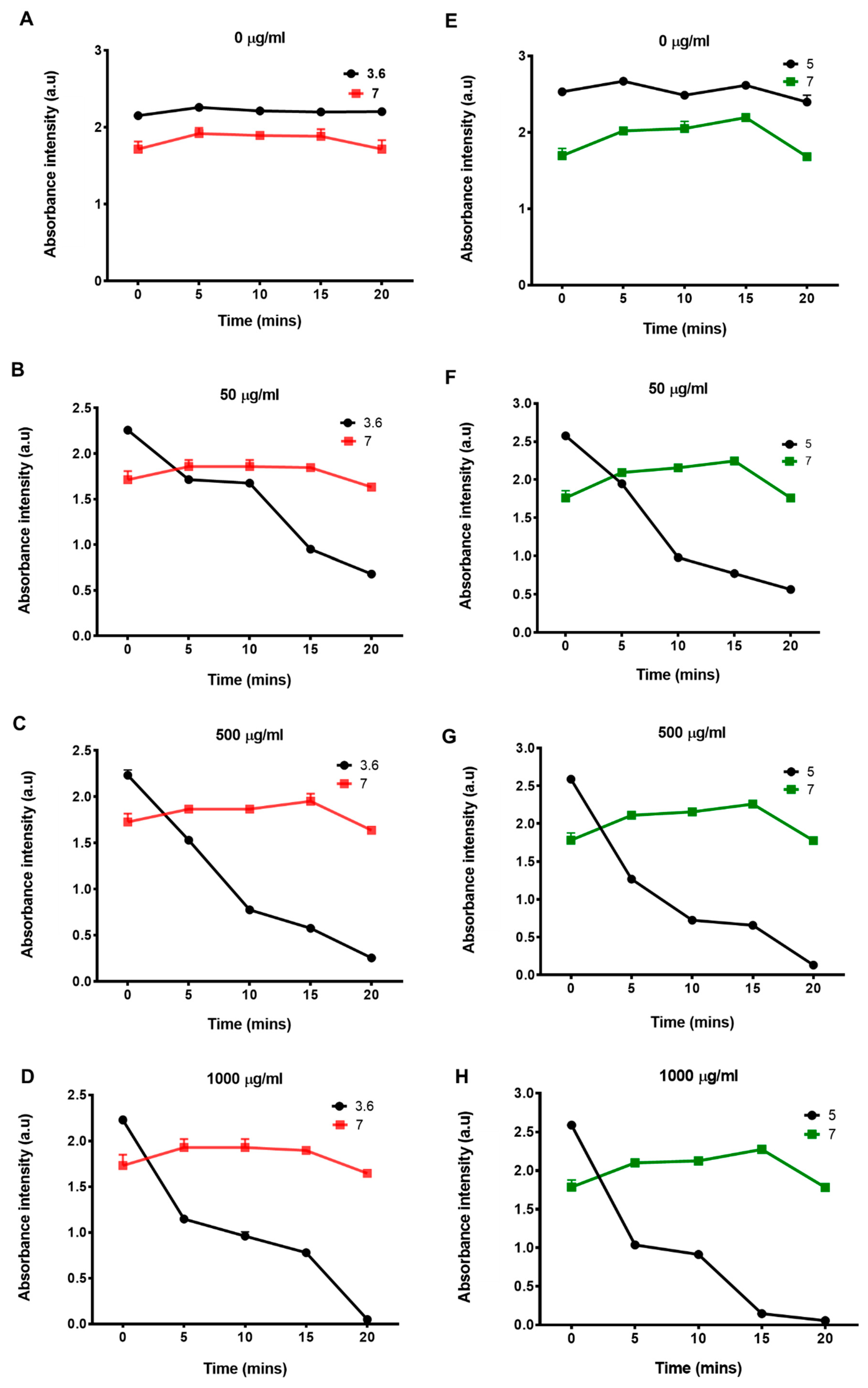
2.2. Basic Characterization of Enzymes
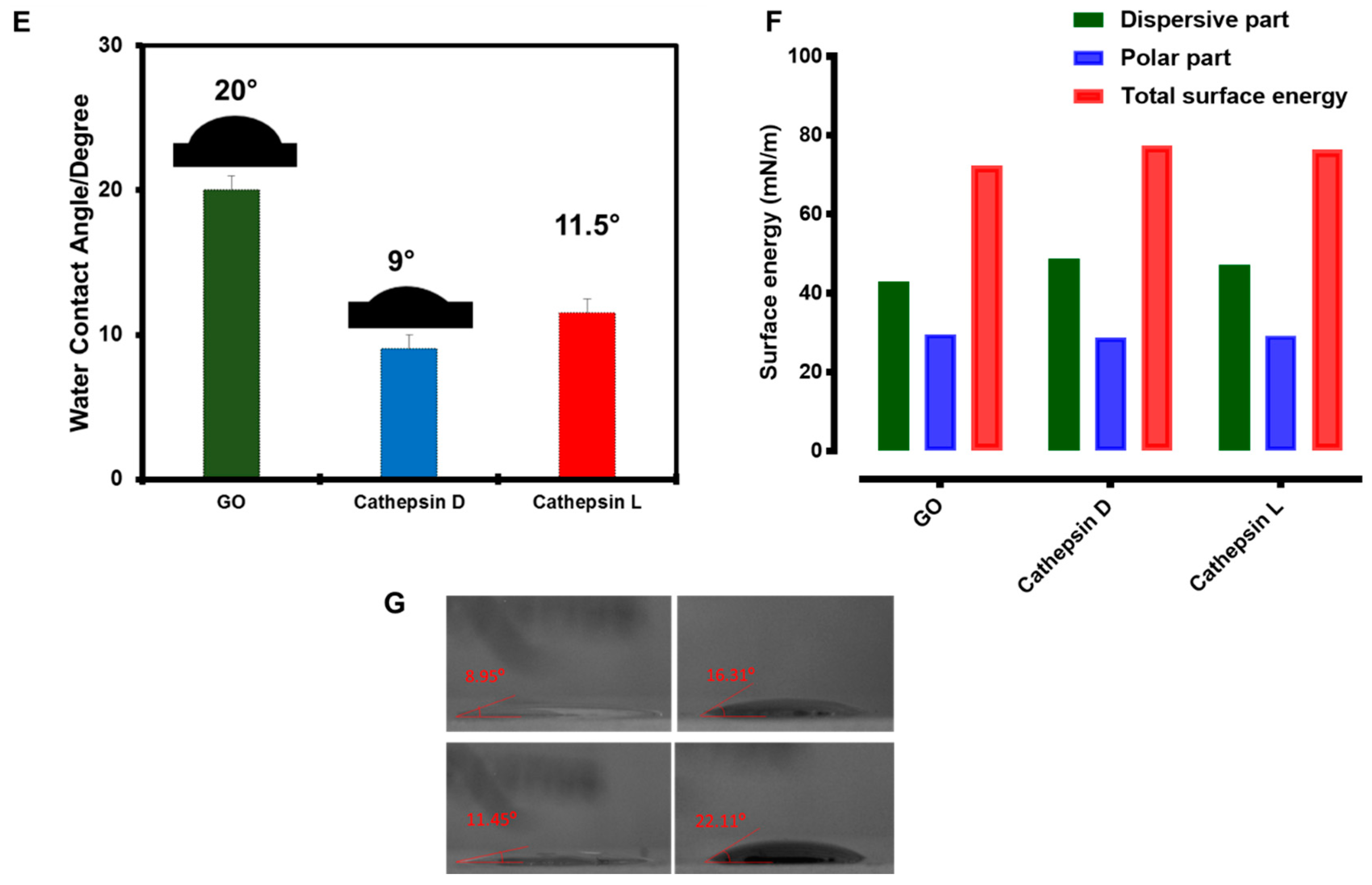
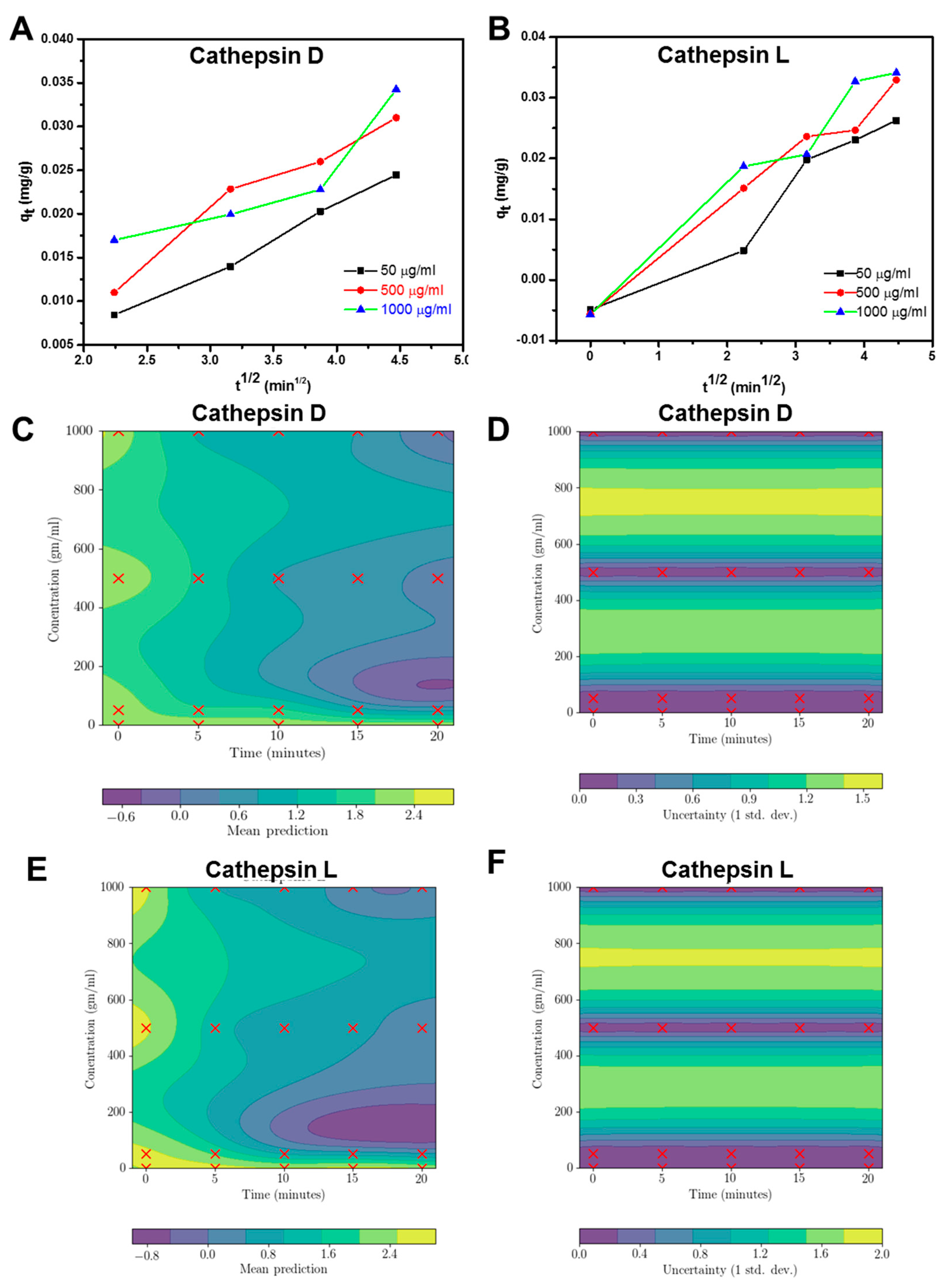
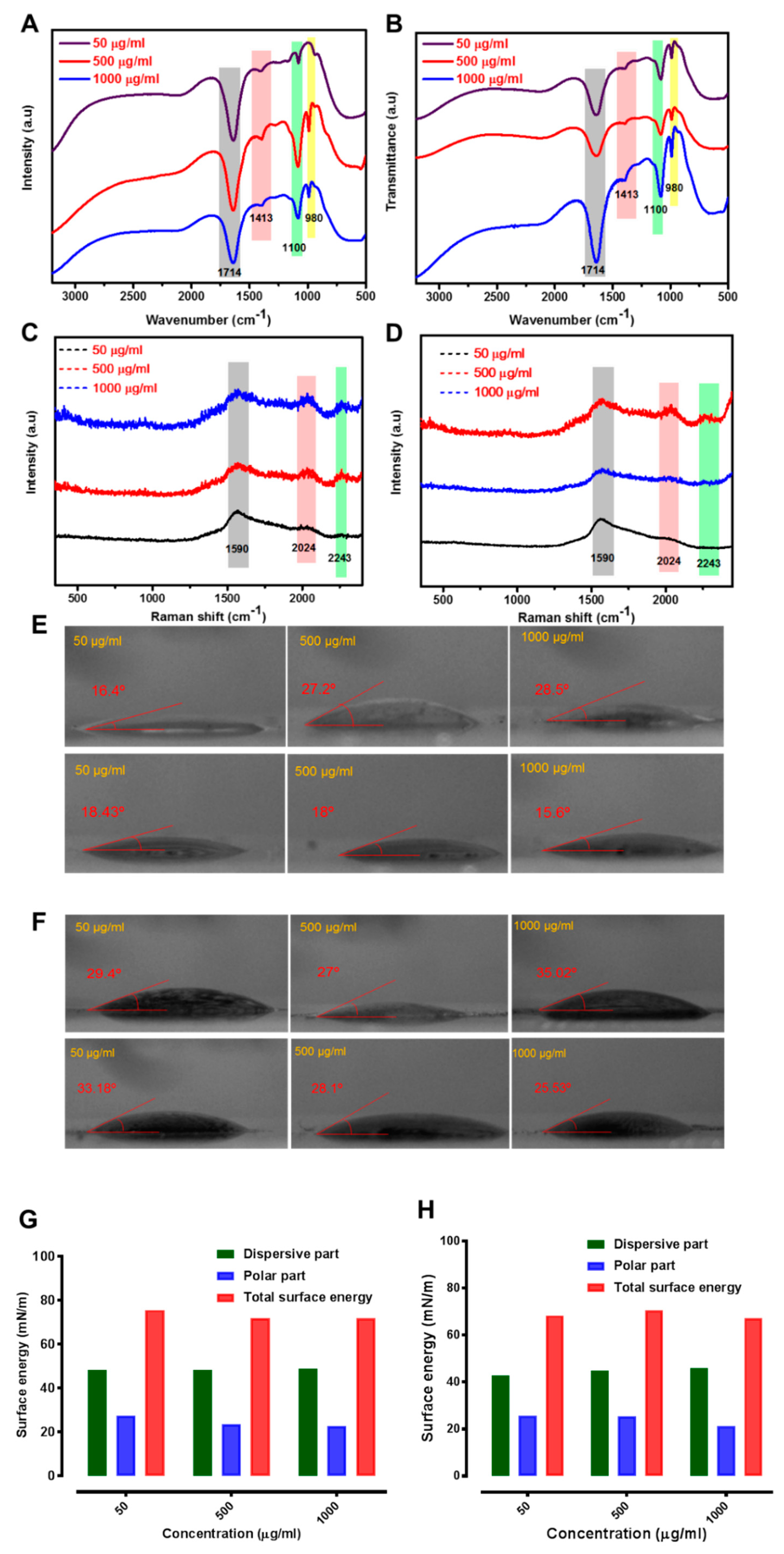
2.3. Enzyme Interaction with GO
3. Discussion
4. Experimental Section
4.1. Synthesis and Characterization
4.2. Cell Viability
4.3. Regression Model
4.4. Water Contact Angle Measurements and Surface Energy Calculations
4.5. Proteolytic Activities
4.6. CathD Experiment
4.7. CathL Experiment
4.8. Enzyme Interaction with GO
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Wang, Q. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Aboagye, E.O.; Ali, S.; Anderson, A.S.; Armes, J.; Berditchevski, F.; Bundred, N.J. Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res. 2013, 15, R92. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, G.M.; Themelis, G.; Crane, L.M.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Bart, J. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-[alpha] targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Aas, T.; Børresen, A.L.; Geisler, S.; Smith-Sørensen, B.; Johnsen, H.; Varhaug, J.E.; Lønning, P.E. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat. Med. 1996, 2, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Doufekas, K.; Olaitan, A. Clinical epidemiology of epithelial ovarian cancer in the UK. Int. J. Women’s Health 2014, 6, 537. [Google Scholar]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Johnson, J. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour–host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein quality control of RNP granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Glondu, M.; Gleizes, M.; Brouillet, J.P.; Vignon, F.; Garcia, M.; Liaudet-Coopman, E. Cathepsin-D affects multiple tumor progression steps in vivo: Proliferation, angiogenesis and apoptosis. Oncogene 2002, 21, 5951. [Google Scholar] [CrossRef] [PubMed]
- Sudhan, D.R.; Rabaglino, M.B.; Wood, C.E.; Siemann, D.W. Cathepsin L in tumor angiogenesis and its therapeutic intervention by the small molecule inhibitor KGP94. Clin. Exp. Metastasis 2016, 33, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.I.; Gutowski, N.; Hannemann, M.; Whatmore, J. The potential role of the proteases cathepsin D and cathepsin L in the progression and metastasis of epithelial ovarian cancer. Biomolecules 2015, 5, 3260–3279. [Google Scholar] [CrossRef] [PubMed]
- Winiarski, B.K.; Wolanska, K.I.; Rai, S.; Ahmed, T.; Acheson, N.; Gutowski, N.J.; Whatmore, J.L. Epithelial ovarian cancer-induced angiogenic phenotype of human omental microvascular endothelial cells may occur independently of VEGF signaling. Transl. Oncol. 2013, 6, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Losch, A.; Schindl, M.; Kohlberger, P.; Lahodny, J.; Breitenecker, G.; Horvat, R.; Birner, P. Cathepsin D in ovarian cancer: Prognostic value and correlation with p53 expression and microvessel density. Gynecol. Oncol. 2004, 92, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bao, K.; Tang, S.; Ai, J.; Hu, H.; Zhang, W. Cyanobacterial peptides as a prototype for the design of cathepsin D inhibitors. J. Pept. Sci. 2017, 23, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Westley, B.R.; May, F.E. Cathepsin D and breast cancer. Eur. J. Cancer 1996, 32A, 15–24. [Google Scholar] [CrossRef]
- Pranjol, M.Z.I.; Gutowski, N.J.; Hannemann, M.; Whatmore, J.L. Cathepsin D non-proteolytically induces proliferation and migration in human omental microvascular endothelial cell via activation of the ERK1/2 and PI3K/AKT pathways. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Steen, H.; Öllinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Pranjol, M.Z.I.; Gutowski, N.J.; Hannemann, M.; Whatmore, J.L. Cathepsin L induces proangiogenic changes in human omental microvascular endothelial cells via activation of the ERK1/2 pathway. Curr. Cancer Drug Targets 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Shi, C.; Yan, Z.; Wu, M. Overexpression of Cathepsin L is associated with chemoresistance and invasion of epithelial ovarian cancer. Oncotarget 2016, 7, 45995. [Google Scholar] [CrossRef] [PubMed]
- Wason, M.; Lu, H.; Yu, L.; Lahiri, S.; Mukherjee, D.; Shen, C.; Zhao, J. Cerium Oxide Nanoparticles Sensitize Pancreatic Cancer to Radiation Therapy through Oxidative Activation of the JNK Apoptotic Pathway. Cancers 2018, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A. Graphene-based materials: The missing piece in nanomedicine? Biochem. Biophys. Res. Commun. 2018, 504, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.N. Graphene and its derivatives as biomedical materials: Future prospects and challenges. Interface Focus 2018, 8, 20170056. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Lin, L.; Ali, M.; Jabeen, F.; Ali, M.; Iqbal, R.; Zhang, S. Investigating the bioavailability of graphene quantum dots in lung tissues via Fourier transform infrared spectroscopy. Interface Focus 2018, 8, 20170054. [Google Scholar] [CrossRef] [PubMed]
- Bugli, F.; Cacaci, M.; Palmieri, V.; Di Santo, R.; Torelli, R.; Ciasca, G.; De Spirito, M. Curcumin-loaded graphene oxide flakes as an effective antibacterial system against methicillin-resistant Staphylococcus aureus. Interface Focus 2018, 8, 20170059. [Google Scholar] [CrossRef] [PubMed]
- Matharu, R.K.; Porwal, H.; Ciric, L.; Edirisinghe, M. The effect of graphene–poly (methyl methacrylate) fibres on microbial growth. Interface Focus 2018, 8, 20170058. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Pranjol, M.Z.I.; Jabeen, F.; Abdullah, T.; Latif, A.; Khalid, A.; Zhang, S. Investigation into the toxic effects of graphene nanopores on lung cancer cells and biological tissues. Appl. Mater. Today 2018, 12, 389–401. [Google Scholar] [CrossRef]
- Tabish, T.A.; Pranjol, M.Z.I.; Karadag, I.; Horsell, D.W.; Whatmore, J.L.; Zhang, S. Influence of luminescent graphene quantum dots on trypsin activity. Int. J. Nanomed. 2018, 13, 1525. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wu, H.; Green, S.J.; Crompton, J.; Zhang, S.; Horsell, D.W. Formation of tunable graphene oxide coating with high adhesion. Phys. Chem. Chem. Phys. 2016, 18, 5086–5090. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, S. Effective solvothermal deoxidization of graphene oxide using solid sulphur as a reducing agent. J. Mater. Chem. 2012, 22, 14385–14393. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, S. Surface energy engineering in the solvothermal deoxidation of graphene oxide. Adv. Mater. Interfaces 2014, 1, 1300078. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, Y.; Chen, L.; Zhu, T.; Ye, K.; Jia, C.; Mo, X. In vitro and in vivo studies of electroactive reduced graphene oxide-modified nanofiber scaffolds for peripheral nerve regeneration. Acta Biomater. 2019, 84, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Memon, F.A.; Gomez, D.E.; Horsell, D.W.; Zhang, S. A facile synthesis of porous graphene for efficient water and wastewater treatment. Sci. Rep. 2018, 8, 1817. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, B.; Park, J.; Yang, J.; Shen, X.; Yao, J. Synthesis of enhanced hydrophilic and hydrophobic graphene oxide nanosheets by a solvothermal method. Carbon 2009, 47, 68–72. [Google Scholar] [CrossRef]
- Tabish, T.A.; Scotton, C.J.J.; Ferguson, D.C.; Lin, L.; der Veen, A.V.; Lowry, S.; Zhang, S. Biocompatibility and toxicity of graphene quantum dots for potential application in photodynamic therapy. Nanomedicine 2018, 13, 1923–1937. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Chabi, S.; Ali, M.; Xia, Y.; Jabeen, F.; Zhang, S. Tracing the bioavailability of three-dimensional graphene foam in biological tissues. Materials 2017, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Pranjol, M.Z.I.; Hayat, H.; Rahat, A.A.; Abdullah, T.M.; Whatmore, J.L.; Zhang, S. In vitro toxic effects of reduced graphene oxide nanosheets on lung cancer cells. Nanotechnology 2017, 28, 504001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, J.; Qiao, C.; Tang, S.; Li, Y.; Yuan, W.; Gao, H. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem. Commun. 2011, 47, 6858–6860. [Google Scholar] [CrossRef] [PubMed]
- Tabish, T.A.; Zhang, S.; Winyard, P.G. Developing the next generation of graphene-based platforms for cancer therapeutics: The potential role of reactive oxygen species. Redox Biol. 2018, 15, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lunova, M.; Smolková, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinová, Š.; Lunov, O. Targeting the mTOR Signaling Pathway Utilizing Nanoparticles: A Critical Overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, J.; Wang, F.; Xiong, Y.; Wu, Y.; Wang, Q.; Liu, S. Improved in vitro and in vivo biocompatibility of graphene oxide through surface modification: Poly (acrylic acid)-functionalization is superior to PEGylation. ACS Nano 2016, 10, 3267–3281. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Song, B.; Liang, H.; Liu, J.; Feng, X.; Deng, B.; Shao, L. Toxicity of graphene-family nanoparticles: A general review of the origins and mechanisms. Part. Fibre Toxicol. 2016, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Volkov, Y.; McIntyre, J.; Prina-Mello, A. Graphene toxicity as a double-edged sword of risks and exploitable opportunities: A critical analysis of the most recent trends and developments. 2d Mater. 2017, 4, 022001. [Google Scholar] [CrossRef]
- Seabra, A.B.; Paula, A.J.; de Lima, R.; Alves, O.L.; Durán, N. Nanotoxicity of graphene and graphene oxide. Chem. Res. Toxicol. 2014, 27, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Morhardt, C.; Ketterer, B.; Heißler, S.; Franzreb, M. Direct quantification of immobilized enzymes by means of FTIR ATR spectroscopy—A process analytics tool for biotransformations applying non-porous magnetic enzyme carriers. J. Mol. Catal. B Enzym. 2014, 107, 55–63. [Google Scholar] [CrossRef]
- Rehman, S.; Movasaghi, Z.; Darr, J.A.; Rehman, I.U. Fourier transform infrared spectroscopic analysis of breast cancer tissues; identifying differences between normal breast, invasive ductal carcinoma, and ductal carcinoma in situ of the breast. Appl. Spectrosc. Rev. 2010, 45, 355–368. [Google Scholar] [CrossRef]
- Movasaghi, Z.; Rehman, S.; Ur Rehman, D.I. Fourier transform infrared (FTIR) spectroscopy of biological tissues. Appl. Spectrosc. Rev. 2008, 43, 134–179. [Google Scholar] [CrossRef]
- Sahoo, J.K.; Sirimuthu, N.M.; Canning, A.; Zelzer, M.; Graham, D.; Ulijn, R.V. Analysis of enzyme-responsive peptide surfaces by Raman spectroscopy. Chem. Commun. 2016, 52, 4698–4701. [Google Scholar] [CrossRef] [PubMed]
- Sée, V.; Free, P.; Cesbron, Y.; Nativo, P.; Shaheen, U.; Rigden, D.J.; Brust, M. Cathepsin L digestion of nanobioconjugates upon endocytosis. ACS Nano 2009, 3, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Berciaud, S.; Cognet, L.; Blab, G.A.; Lounis, B. Photothermal Heterodyne Imaging of Individual Non-fluorescent Nanoclusters and Nanocrystals. Phys. Rev. Lett. 2004, 93, 257402. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, C.E. Gaussian Processes in Machine Learning. In Summer School on Machine Learning; Springer: Berlin, Germany, 2003; pp. 63–71. [Google Scholar]
- Fowkes, F.M. Attractive Forces at Interfaces. Ind. Eng. Chem. 1964, 56, 40. [Google Scholar] [CrossRef]
- Malana, M.A.; Qureshi, R.B.; Ashiq, M.N. Adsorption studies of arsenic on nano aluminium doped manganese copper ferrite polymer (MA, VA, AA) composite: Kinetics and mechanism. Chem. Eng. J. 2011, 172, 721. [Google Scholar] [CrossRef]
- Alkan, M.; Demirbaş, Ö.; Celikcapa, S.; Doğan, M. Sorption of acid red 57 from aqueous solution onto sepiolite. J. Hazard. Mater. 2004, 116, 135. [Google Scholar] [CrossRef] [PubMed]
- Altenor, S.; Carene, B.; Emmanuel, E.; Lambert, J.; Ehrhardt, J.J.; Gaspard, S. Adsorption studies of methylene blue and phenol onto vetiver roots activated carbon prepared by chemical activation. J. Hazard. Mater. 2009, 165, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Xu, Z. Lead sorption from aqueous solutions on chitosan nanoparticles. Colloids Surf. A Physicochem. Eng. Aspects 2004, 251, 183–190. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabish, T.A.; Pranjol, M.Z.I.; Horsell, D.W.; Rahat, A.A.M.; Whatmore, J.L.; Winyard, P.G.; Zhang, S. Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L As A Novel Anti-Metastatic Enzyme Cancer Therapy. Cancers 2019, 11, 319. https://doi.org/10.3390/cancers11030319
Tabish TA, Pranjol MZI, Horsell DW, Rahat AAM, Whatmore JL, Winyard PG, Zhang S. Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L As A Novel Anti-Metastatic Enzyme Cancer Therapy. Cancers. 2019; 11(3):319. https://doi.org/10.3390/cancers11030319
Chicago/Turabian StyleTabish, Tanveer A., Md Zahidul I. Pranjol, David W. Horsell, Alma A. M. Rahat, Jacqueline L. Whatmore, Paul G. Winyard, and Shaowei Zhang. 2019. "Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L As A Novel Anti-Metastatic Enzyme Cancer Therapy" Cancers 11, no. 3: 319. https://doi.org/10.3390/cancers11030319
APA StyleTabish, T. A., Pranjol, M. Z. I., Horsell, D. W., Rahat, A. A. M., Whatmore, J. L., Winyard, P. G., & Zhang, S. (2019). Graphene Oxide-Based Targeting of Extracellular Cathepsin D and Cathepsin L As A Novel Anti-Metastatic Enzyme Cancer Therapy. Cancers, 11(3), 319. https://doi.org/10.3390/cancers11030319