Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of CAFs in PDAC
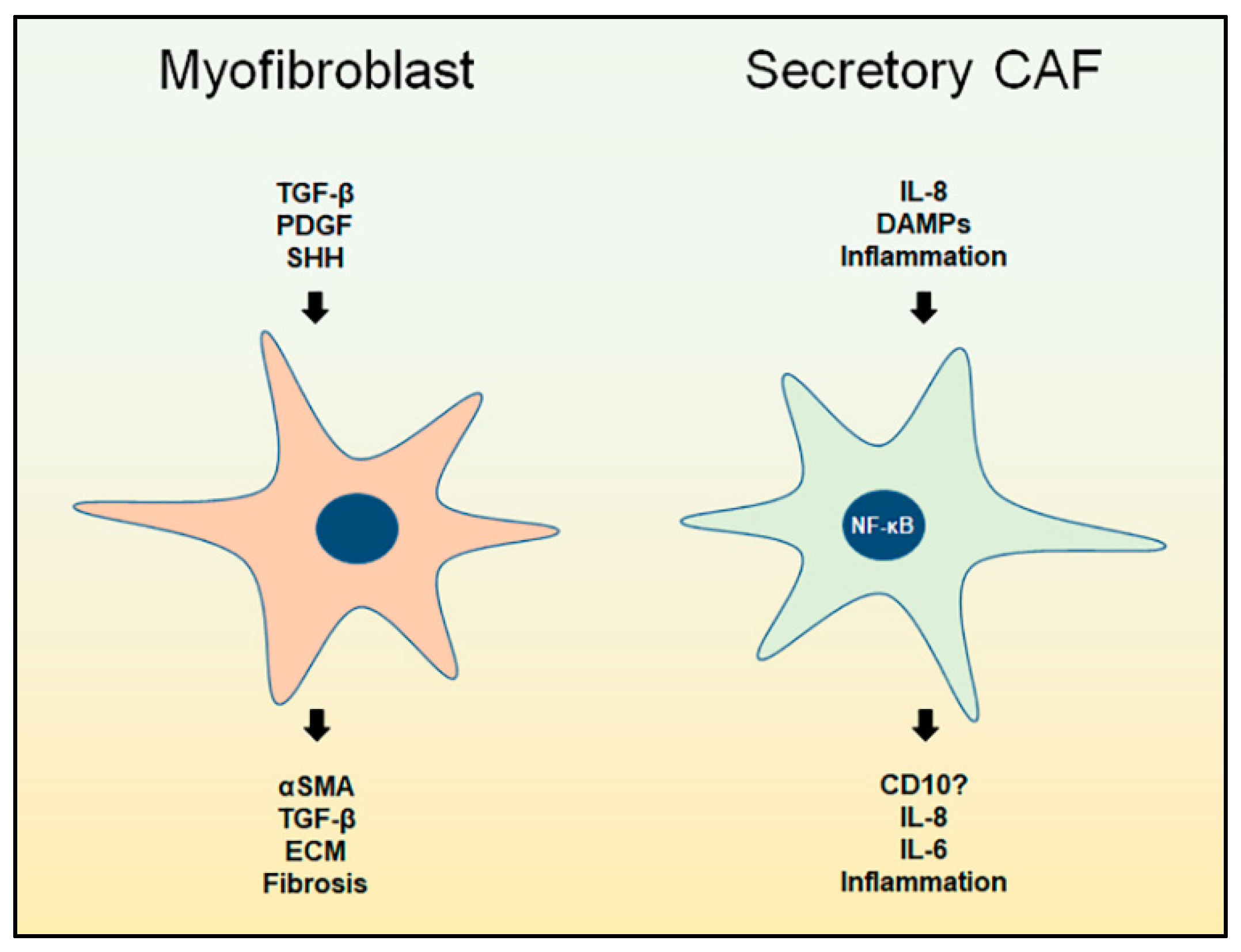
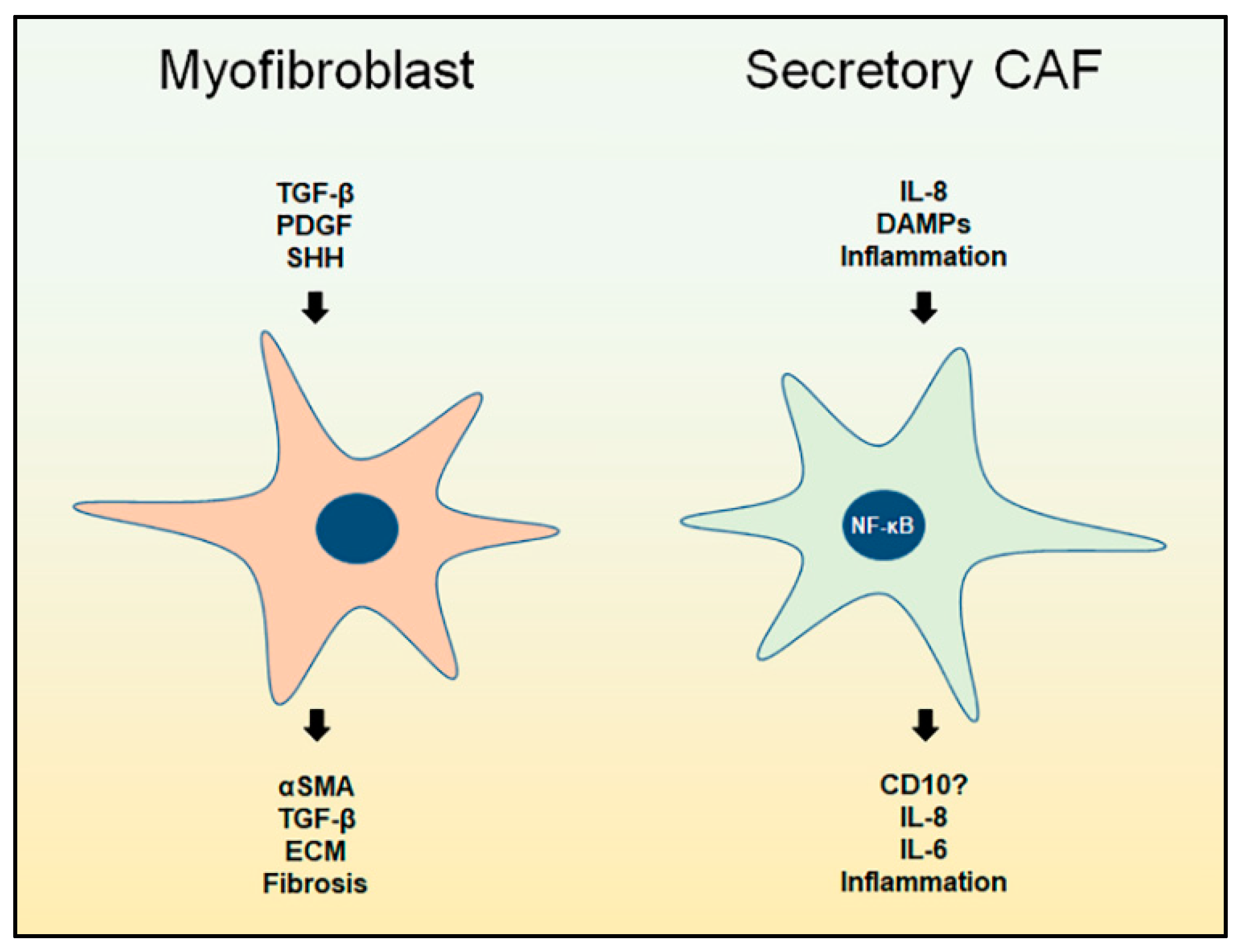
3. Myofibroblast CAFs
3.1. Overview
3.2. Wound Healing
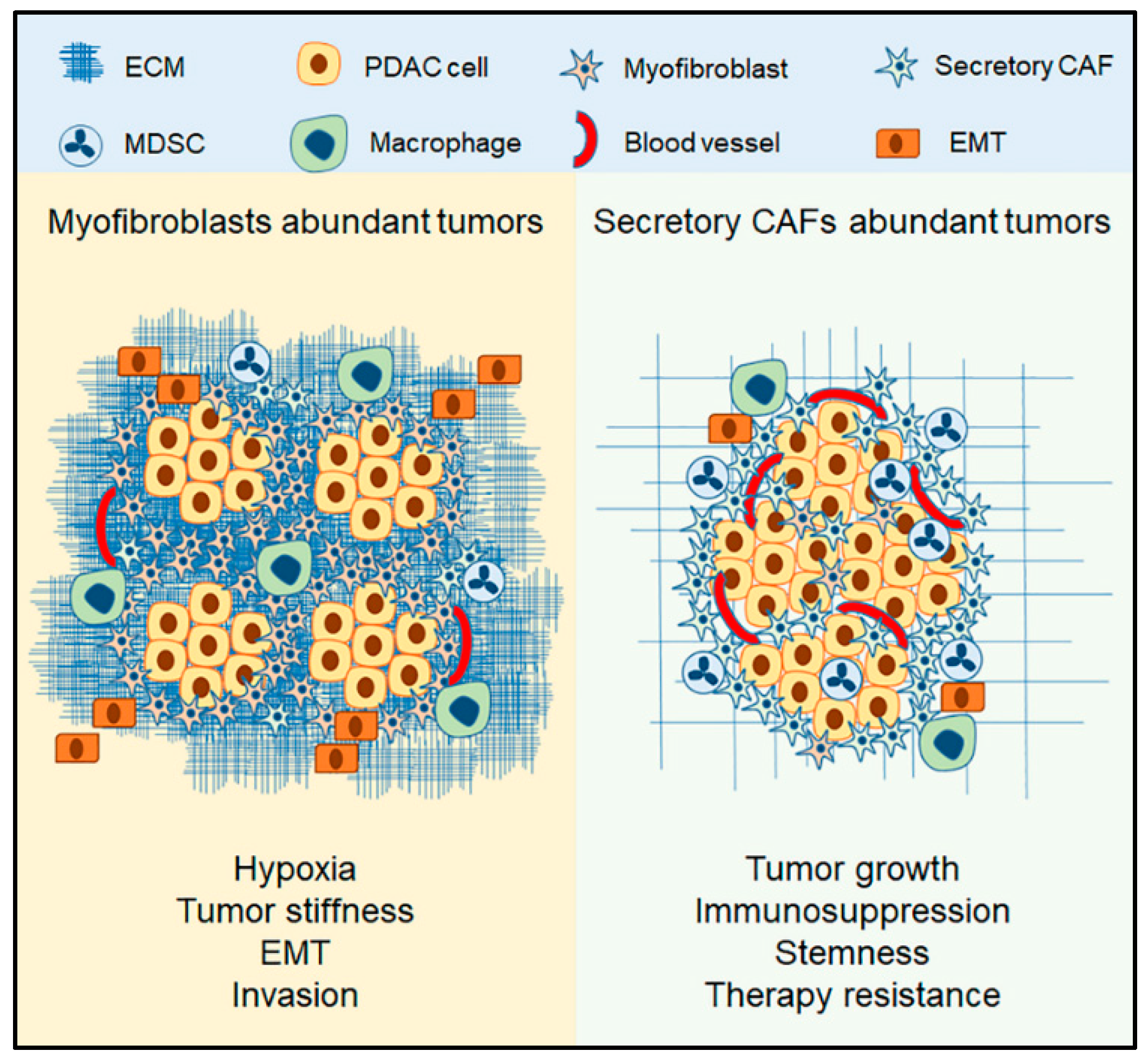
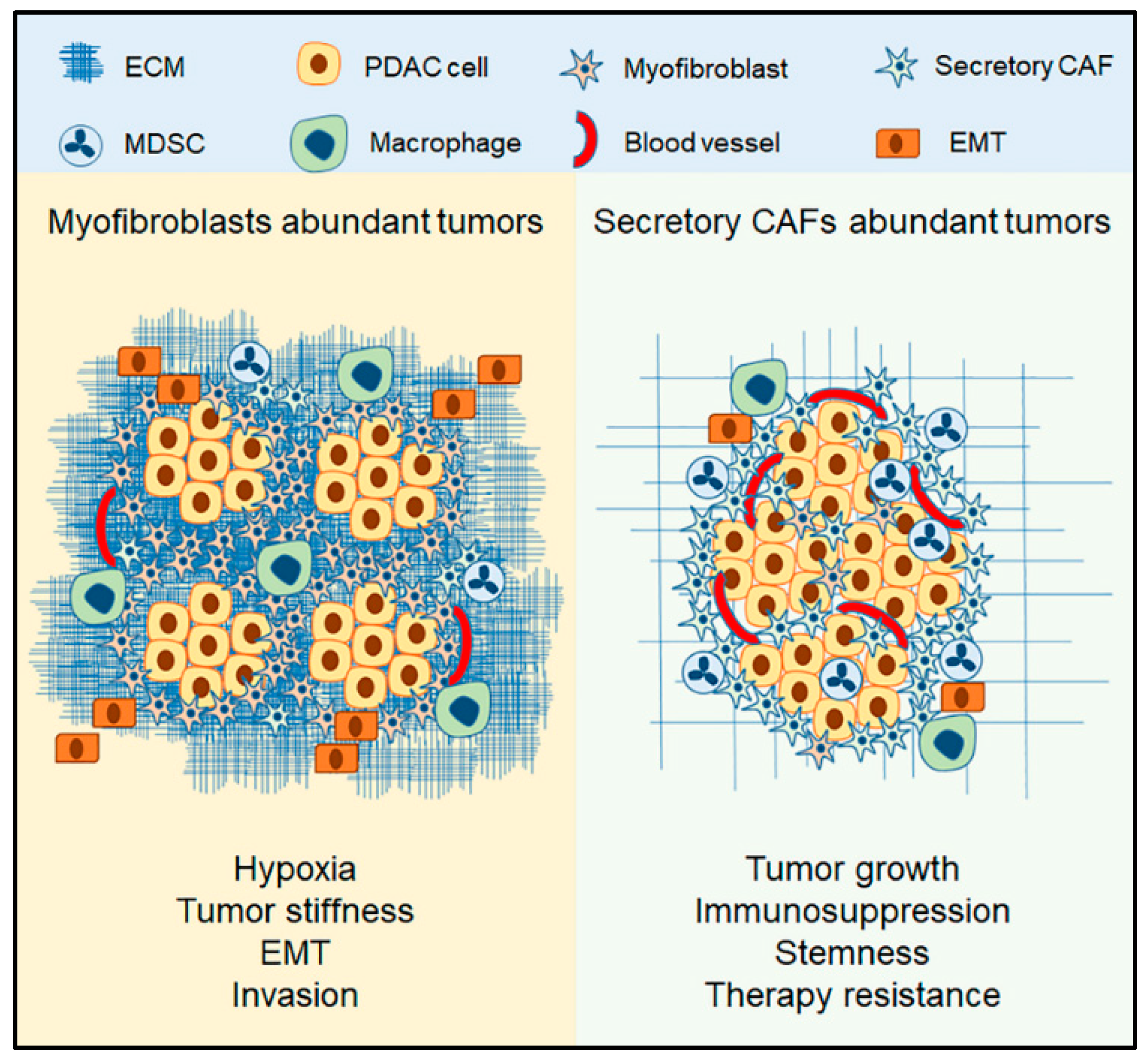
3.3. The Context of Myofibroblasts in PDAC
4. Secretory CAFs
4.1. Overview
4.2. CAFs and Inflammation
4.3. The Context of Secretory CAFs
4.4. The Role Secretory CAFs in PDAC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Moir, J.A.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic Reaction in Pancreatic Cancer. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S. Mechanisms of pancreatic fibrosis. Dig. Dis. 2004, 22, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic Cancer. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Kleeff, J.; Beckhove, P.; Esposito, I.; Herzig, S.; Huber, P.E.; Lohr, J.M.; Friess, H. Pancreatic cancer microenvironment. Int. J. Cancer 2007, 121, 699–705. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, R.A.; Chang, H.; Dumont, N.; Rabban, J.T.; Chen, Y.-Y.; Fontenay, G.V.; Berman, H.K.; Gauthier, M.L.; Zhao, J.; Hu, D.; et al. CD36 Repression Activates a Multicellular Stromal Program Shared by High Mammographic Density and Tumor Tissues. Cancer Discov. 2012, 2, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Vierkant, R.A.; Frank, R.D.; Winham, S.; Visscher, D.W.; Pankratz, V.S.; Scott, C.G.; Brandt, K.; Sherman, M.E.; Radisky, D.C.; et al. Association between mammographic breast density and histologic features of benign breast disease. Breast Cancer Res. 2017, 19, 134. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Baba, T.; Shinagawa, K.; Matsushima, K.; Mukaida, N. Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int. J. Cancer 2014, 135, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer Microenvironment and Inflammation: Role of Hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2012, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; de Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma-Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, H.; Webster, M.R.; Behera, R.; Jimenez Valencia, A.M.; Wirtz, D.; Weeraratna, A.T.; Shenoy, V.B. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proc. Natl. Acad. Sci. USA 2017, 114, E1617–E1626. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.; Fmj, W.; Slf, P.; Chance, V.; Williams, E.J.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P.; Fine, D.R. The Role of Transforming Growth Factor-β in the Regulation of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases by Pancreatic Stellate Cells: Implications for Chronic Pancreatitis. Clin. Sci. 2002, 103, 40P–41P. [Google Scholar] [CrossRef]
- Shek, F.W.-T.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.F.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of Transforming Growth Factor-β1 by Pancreatic Stellate Cells and Its Implications for Matrix Secretion and Turnover in Chronic Pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Williams, L.V.; Schwarzbauer, J.E. Tissue repair and the dynamics of the extracellular matrix. Int. J. Biochem. Cell Biol. 2004, 36, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, S.; Farooq, S.M.; Castelvetere, M.P.; Hou, Y.; Gao, J.-L.; Navarro, J.V.; Oupicky, D.; Sun, F.; Li, C. A Chemokine Receptor CXCR2 Macromolecular Complex Regulates Neutrophil Functions in Inflammatory Diseases. J. Biol. Chem. 2011, 287, 5744–5755. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.; Barron, L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Katuri, V.; Srinivasan, R.; Fogt, F.; Redman, R.; Anand, G.; Said, A.; Fishbein, T.; Zasloff, M.; Reddy, E.P.; et al. Transforming Growth Factor-β Suppresses Nonmetastatic Colon Cancer through Smad4 and Adaptor Protein ELF at an Early Stage of Tumorigenesis. Cancer Res. 2005, 65, 4228–4237. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pouponnot, C.; Massague, J. Dual role of the Smad4/DPC4 tumor suppressor in TGFβ-inducible transcriptional complexes. Genes Dev. 1997, 11, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2014, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 Gene Mutations Are Associated with Poor Prognosis in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srinivasan, R.; Wig, J.D. SMAD4 Genetic Alterations Predict a Worse Prognosis in Patients with Pancreatic Ductal Adenocarcinoma. Pancreas 2012, 41, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Tascilar, M.; Skinner, H.G.; Rosty, C.; Sohn, T.; Wilentz, R.E.; Offerhaus, G.J.; Adsay, V.; Abrams, R.A.; Cameron, J.L.; Kern, S.E.; et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2001, 7, 4115–4121. [Google Scholar] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Valyi-Nagy, I.; Heldin, C.H.; Herlyn, M.; Westermark, B. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl. Acad. Sci. USA 1993, 90, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.M.; Sizemore, G.M.; Shukla, V.C.; Avendano, A.; Sizemore, S.T.; Chang, J.J.; Kladney, R.D.; Cuitiño, M.C.; Thies, K.A.; Verfurth, Q.; et al. Stromal PDGFR-α Activation Enhances Matrix Stiffness, Impedes Mammary Ductal Development, and Accelerates Tumor Growth. Neoplasia 2017, 19, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Kayed, H.; Kleeff, J.R.; Osman, T.; Keleg, S.; Büchler, M.W.; Friess, H. Hedgehog Signaling in the Normal and Diseased Pancreas. Pancreas 2006, 32, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008, 16, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Raguz, S.; Gil, J. Control of senescence by CXCR2 and its ligands. Cell Cycle 2008, 7, 2956–2959. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Hinds, P.; Pietruska, J. Senescence and tumor suppression. F1000Res. 2017, 6, 2121. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S. Stromal-epithelial interactions in aging and cancer: Senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The Gene Expression Program of Prostate Fibroblast Senescence Modulates Neoplastic Epithelial Cell Proliferation through Paracrine Mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2017, 15, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Korc, M. RelA: A tale of a stitch in time. J. Clin. Investig. 2016, 126, 2799–2801. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Wormann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwachter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 2016, 126, 2919–2932. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. (Ed.) Nuclear Factor-KappaB. In Encyclopedia of Cancer; Springer: Berlin/Heidelberg, Germany, 2017; p. 3151. [Google Scholar]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Maguer-Satta, V.; Besancon, R.; Bachelard-Cascales, E. Concise review: Neutral endopeptidase (CD10): A multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011, 29, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Iwaya, K.; Izumi, M.; Kuroda, M.; Serizawa, H.; Koyanagi, Y.; Mukai, K. Expression of CD10 by stromal cells during colorectal tumor development. Hum. Pathol. 2002, 33, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, K.; Ogawa, H.; Izumi, M.; Kuroda, M.; Mukai, K. Stromal expression of CD10 in invasive breast carcinoma: A new predictor of clinical outcome. Virchows Arch. 2002, 440, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.B. CD10-positive Stromal Cells in Gastric Carcinoma: Correlation with Invasion and Metastasis. Jpn. J. Clin. Oncol. 2005, 35, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ Pancreatic Stellate Cells Enhance the Progression of Pancreatic Cancer. Gastroenterology 2010, 139, 1041–1051.e8. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Beccari, A.R.; Bertini, R.; Cavicchia, M.R.; Giorgini, S.; Allegretti, M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 2006, 112, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Burdick, M.D.; Mestas, J.; Gomperts, B.; Keane, M.P.; Belperio, J.A. Cancer CXC chemokine networks and tumour angiogenesis. Eur. J. Cancer 2006, 42, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Varney, M.; Rachagani, S.; Ouellette, M.M.; Batra, S.K.; Singh, R.K. CXCR2 signaling regulates KRAS(G(1)(2)D)-induced autocrine growth of pancreatic cancer. Oncotarget 2016, 7, 7280–7296. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A. The Role of CXCR2 in Pancreatic Cancer Development and Progression. Ph.D. Thesis, University of Nebraska Medical Center, Omaha, NE, USA, 2015. [Google Scholar]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Awaji, M.; Varney, M.; Purohit, A.; Batra, S.; Singh, R. CXCR2 signaling in pancreatic cancer induces a tumor supporting inflammatory phenotype of the cancer-associated fibroblasts. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Awaji, M.; Varney, M.; Purohit, A.; Batra, S.K.; Singh, R.K. CXCR2 acts as a checkpoint regulator of the pancreatic stellate cells activity within pancreatic cancer tumor microenvironment. In Proceedings of the AACR Annual Meeting 2017, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Acosta, J.C.; Gil, J. A role for CXCR2 in senescence, but what about in cancer? Cancer Res. 2009, 69, 2167–2170. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Biavasco, R.; Borgiani, B.; Dagna, L. Oncogene-induced senescence as a new mechanism of disease: The paradigm of erdheim-chester disease. Front. Immunol. 2014, 5, 281. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Varney, M.; Singh, R.K. Host CXCR2-Dependent Regulation of Melanoma Growth, Angiogenesis, and Experimental Lung Metastasis. Cancer Res. 2009, 69, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow–derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awaji, M.; Singh, R.K. Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers 2019, 11, 290. https://doi.org/10.3390/cancers11030290
Awaji M, Singh RK. Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers. 2019; 11(3):290. https://doi.org/10.3390/cancers11030290
Chicago/Turabian StyleAwaji, Mohammad, and Rakesh K. Singh. 2019. "Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma" Cancers 11, no. 3: 290. https://doi.org/10.3390/cancers11030290
APA StyleAwaji, M., & Singh, R. K. (2019). Cancer-Associated Fibroblasts’ Functional Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancers, 11(3), 290. https://doi.org/10.3390/cancers11030290